在呼吸链中,NADH氧化与离子跨膜移位相结合,形成电化学梯度。在人类病原体霍乱弧菌中,钠泵NADH:醌氧化还原酶(Na+-NQR)通过一种迄今未知的机制产生钠梯度。我们发现Na+-NQR中的离子泵送是由电子转移到离子移位的大构象变化驱动的。我们已经确定了一系列的低温电镜和x射线结构的Na+-NQR,代表催化循环的快照。Na+-NQR的六个亚基NqrA, B, C, D, E和F包含一组独特的辅助因子,这些辅助因子将电子从NADH穿过膜两次运送到醌。独特的膜内[2Fe-2S]簇的氧化还原状态协调亚基NqrC的运动,NqrC充当电子转移开关。我们认为这种开关运动控制了Na+从亚基NqrB的结合位点的释放。
在线粒体和细菌的呼吸链中,NADH的氧化和泛醌的还原与离子易位和电化学梯度的建立有关。该反应由络合物I或Na+-NQR催化,但这两种膜蛋白复合物具有完全不同的结构。值得注意的是,Na+-NQR仅发生在细菌中,并广泛存在于霍乱弧菌和耐多药假单胞菌和克雷伯氏菌等病原体中,使其成为新抗生素的有希望的靶点。Na+-NQR具有一组独特的辅助因子s2(一个黄素腺嘌呤二核苷酸(FAD),两个共价结合的黄素单核苷酸(FMNs),一个核黄素和两个铁硫中心),并偶联NADH:泛素氧化还原,使两个Na+离子在细胞质膜上易位e3。我们小组的3.5-?-resolution x射线结构揭示了Na+-NQR的六个亚基的排列和拓扑结构。然而,Na+-NQR的机制仍然不清楚,因为该结构只提供了催化循环的一种状态的快照。这种x射线结构和最近的冷冻电镜结构的分辨率有限,不能分辨水分子或离子,侧链和辅助因子核黄素的位置也不能明确地分辨出来。本文报道的高分辨率低温电镜结构揭示了所有辅助因子的精确定位和性质,包括NqrD和NqrE亚基中的[2Fe-2S]簇。此外,在NqrB中还鉴定出两个钠离子。
利用低温电子显微镜(cryo-EM)和x射线晶体学测定了不同构象的Na+-NQR的结构。在2.1至3.2 ?的分辨率下,测定了Na+-NQR在天然状态下、与泛素-1 (UQ-1)、泛素-2 (UQ-2)、NADH和泛素-2、抑制剂2-十烷基-4-喹唑啉胺(DQA)或抑制剂2-庚基-4-羟基喹啉n-氧化物(HQNO)配合物下的6种低温电镜结构(表1和扩展数据图1和2)。此外,我们用新模型改进了x射线结构,并确定了DQA处理状态下的x射线结构。这些结构由NqrF的可溶性结构域及其底物NADH结合的高分辨率结构补充(表2)。
表1 Cryo-EM数据收集、细化和验证统计
表2 x射线数据收集、细化和验证的统计数据
Na+-NQR由三个完整的膜亚基NqrB、NqrD和NqrE以及三个亲水性亚基NqrA、NqrC和NqrF组成。NqrA和NqrF突出在细胞质中,而NqrC位于细胞质外周(图1)。NqrC和NqrF通过单个跨膜螺旋锚定在膜中,而NqrA缺乏跨膜螺旋,与NqrB紧密结合。分析了几种与NqrB、NqrD和NqrE结合的脂质和洗涤剂分子(扩展数据图3)。
图1:Na的cryo-EM和x射线结构比较+-NQR。
a,用低温电镜测定Na+-NQR与NADH和泛素-2复合物的整体结构。b, x射线晶体学测定的Na+-NQR结构。这两种结构的不同之处在于NqrC的质周亲水结构域的位置。c, NqrF与NADH和UQ-2结合的cryo-EM(红色)和x射线(灰色)结构叠加。NqrD(洋红色)和NqrE(青色)为表面。与x射线结构相比,在低温电镜结构中,NqrF的fnr样结构域被侧向移动,而铁氧还蛋白(Fdxn)样结构域向膜亚基方向旋转。d, cryo-EM测定Na+-NQR中氧化还原辅因子的边沿距离。e,通过x射线晶体学测定的Na+-NQR中氧化还原辅因子的边沿距离。灰色条表示膜的位置。
NqrF亚基由两个结构域组成,一个是氨基端[2Fe-2S]类铁氧还蛋白和一个羧基端fad结合结构域,类似于铁氧还蛋白- nadph还原酶(FNRs)。这两个结构域由一个柔性连接体连接,并通过一个单n端跨膜螺旋锚定在膜上。c端fnr样结构域催化NADH氧化,电子通过铁氧化还蛋白样结构域转移到完整的膜亚基NqrD和NqrE上。(NqRD-E)。我们确定了Na+-NQR在NADH和UQ-2存在下的低温电镜结构,分辨率为2.86 ?,通过密度修饰将其提高到2.55 ?。结合的NADH在图中清晰可见,烟酰胺Π-stacked平行于FAD的异alloxazine,以实现有效的氢化物转移(扩展数据图4a,b)。通过NqrF分离的fnr样结构域的x射线结构,以及缺乏对NADH结合很重要的c -末端F406的NqrF变体,进一步表征了NADH的结合模式(扩展数据图4c,d)。
在本文报道的cro - em和x射线结构中,NqrF的铁氧化还原蛋白样结构域位于不同的位置(图1和补充图1),说明该结构域在跨膜螺旋和fnr样结构域之间灵活连接。这使我们能够推断该结构域的构象变化(补充图1a),并模拟其在催化循环中的运动(补充视频1)。该模型预测,类铁氧化还原蛋白结构域的[2Fe-2S]簇与膜亚基NqrD-E中的[2Fe-2S]簇接近15 ?。为了证实NqrF的这些大构象运动,我们进行了化学交联并通过质谱法监测了交联(补充数据集1和2)。
先前x射线结构的分辨率太低,无法确定NqrD-E亚基中先前确定的氧化还原中心的确切性质。这两个亚基极有可能源于基因复制,并呈现出倒置的拓扑结构,在两个亚基的中心附近有四个保守的Cys残基。异常密度差表明存在一个以上的铁原子。khikawa等人最近的一项低温电镜研究报告称,NqrD-E中可能存在[2Fe-2S]簇;然而,没有提供实验证据。我们使用一套全面的光谱技术来表征膜中的[2Fe-2S]簇。高分辨率结构、光谱数据、动力学和分析数据表明,NqrD-E在膜中央配位了一个[2Fe-2S]簇,尽管这个簇以前没有被光谱技术检测到。我们生成了缺乏[2Fe-2S]NqrF (NqrF- c70a)或[2Fe-2S]NqrD-E (NqrD-C29A)的变体(补充表1)。通过圆二色性(CD)、电子顺磁共振(EPR)、57Fe M?ssbauer和Fe Kα高能荧光检测(HERFD) x射线吸收光谱(XAS)分析了野生型Na+-NQR和变体。在没有[2Fe-2S]NqrF的变体中,我们观察到位于NqrD-E亚基的[2Fe-2S]簇的光谱特征(图2a,b)。该团簇的CD光谱显示了在紫外可见区域的跃迁,这是[2Fe-2S]铁氧化还原蛋白的典型特征(图2c)。膜内[2Fe-2S]NqrD-E的EPR谱在g=~2.01 (g是各向异性g张量的一个分量)处表现出明显的特征,与自由基信号一致;仅在高功率设置下观察到g=~1.94的特征,表明这是一个快速弛豫的[2Fe-2S]簇。通过两个分量的模拟可以充分再现EPR光谱:g=~2.01的各向同性自由基和g||=2.02和g⊥=1.94的主导轴向分量(图2f和补充图2b-e)。[2Fe-2S]NqrD-E团簇的零场80 K M?ssbauer光谱证实了这些结果,显示出不对称双重态,异构体位移为0.3 mm s - 1,四极分裂为0.51 mm s - 1(图2e)。[2Fe-2S]NqrD-E星团的EXAFS显示典型的Fe-Fe距离为2.70 ?, Fe-S距离为2.28 ?(图2d和补充图3)。
图2:NqrD-E含有催化跨膜电子转移的[2Fe-2S]簇。
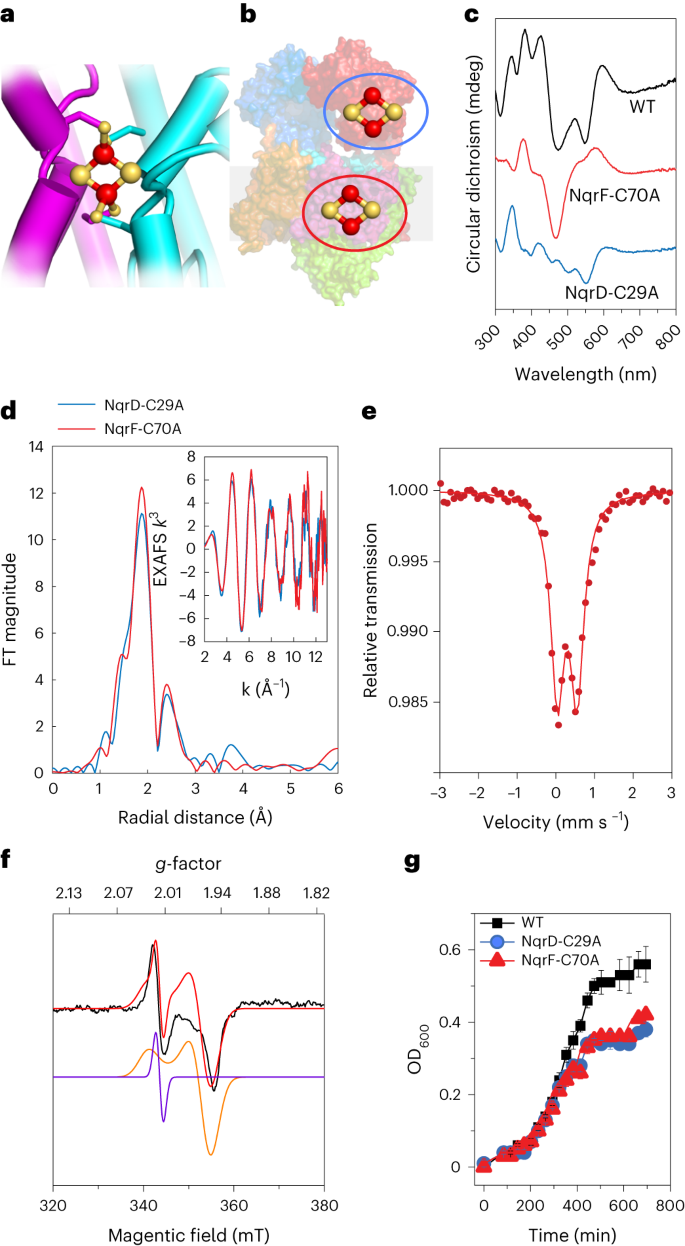
a,亚基NqrD-E协调[2Fe-2S]簇的特写视图,半胱氨酸来自两个亚基。b,胞质NqrF和膜亚基NqrD-E中[2Fe-2S]簇的定位。蓝色圆圈表示[2Fe-2S]NqrF簇;红圈表示[2Fe-2S]NqrD-E星团。c,野生型(WT) Na+-NQR的CD谱;NqrF- c70a,不含[2Fe-2S]NqrF;NqrD-C29A,不含[2Fe-2S]NqrD-E。图中显示了WT Na+-NQR(黑色迹线)、NqrF- c70a中的[2Fe-2S]NqrD-E星团(红色迹线)和NqrD-C29A中的[2Fe-2S]NqrF星团(蓝色迹线)的光谱。d, NqrF- c70a中的[2Fe-2S]NqrF - e团簇(红色)和NqrD-C29A中的[2Fe-2S]NqrF团簇(蓝色)的k3加权EXAFS和傅里叶变换光谱。e, NqrF-C70A中[2Fe-2S]NqrD-E星团的57Fe M?ssbauer光谱。f, Na+-NQR变体NqrF-C70A的10 K x波段EPR谱显示[2Fe-2S]NqrD-E簇,用两个组分模拟:[2Fe-2S]簇(橙色;g||=2.02, g⊥=1.94,线宽=50g)和一个根状信号(紫色;giso=2.01,线宽=20g,相对重量4%)。g,表达WT Na+-NQR(黑色)的霍乱弧菌生长曲线,或变异NqrF- c70a中含有[2Fe-2S]NqrD-E簇(红色)的Na+-NQR变体,或变异NqrD-C29A中含有[2Fe-2S]NqrF簇(蓝色)的Na+-NQR变体。OD600, 600 nm光密度。增长数据以平均值±s.d表示。, n=3个生物独立样本。
源数据
电子从[2Fe-2S]NqrD-E转移到位于外质中的FMNNqrC(图1)。在之前的x射线结构中,NqrC与NqrD-E亚基相互作用,FMNNqrC深埋在NqrD-E中,靠近膜内的[2Fe-2S]NqrD-E。FMNNqrC与[2Fe-2S]NqrD-E之间的7.2-?距离促进了电子转移,而在x射线结构中观察到的FMNNqrC与FMNNqrB之间的~22 ?距离阻碍了电子转移6 (Supplementary Table 4)。我们提出NqrC发生了很大的构象变化以允许电子转移2。本文报道的低温电镜结构和Kishikawa等人最近的低温电镜研究证实了这一点,他们都显示NqrC位于NqrB(图3)。与x射线结构相比,NqrC在低温电镜结构中的位置对应于26°的旋转运动和21.6°的最大平移运动?。在cryo-EM结构中,FMNNqrC与FMNNqrB的距离为5.7 ?,与非常快的电子转移相适应(Supplementary Table 4)。NqrC的这种构象变化在Na+-NQR中起着开关作用,弥补了NqrD-E中氧化还原中心与NqrB之间较大的距离。有趣的是,当晶体与抑制剂DQA孵育时,我们在x射线结构中观察到NqrC的这种开关运动。通过在NqrB或NqrD-E上通过工程二硫键锁定NqrC的变体证实了NqrC的开关(图3和扩展数据图5)。
图3:NqrC作为公司信息电子转移开关。
Na+-NQR在两种状态下的结构对位,突出了NqrC在低温电镜和x射线结构中的两种构象。在低温电镜结构中发现的NqrC位于NqrB,用绿色表示。位于NqrD-E的NqrC,在x射线结构中检测到,以灰色显示。膜平面用灰色条表示。NqrC的运动对应于亲水结构域的26度旋转。
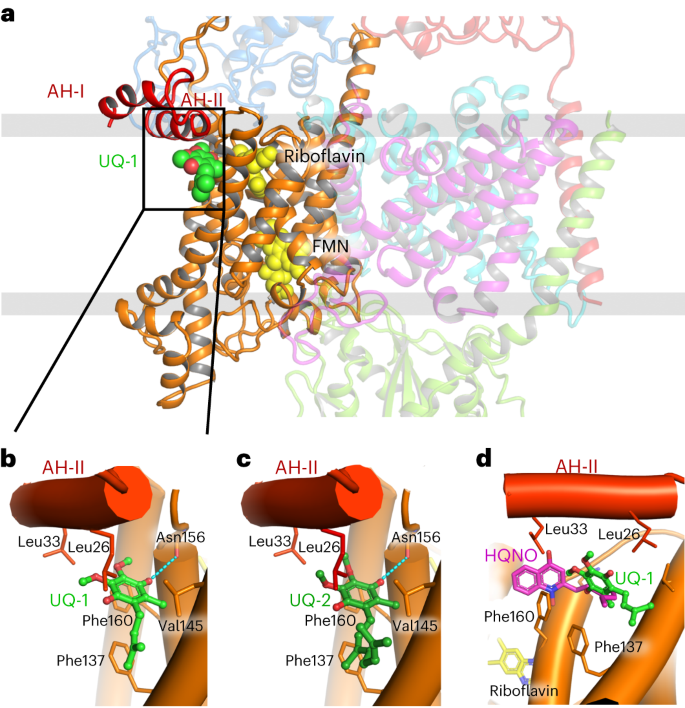
在NqrB中,电子从FMNNqrB转移到核黄素NqrB。如Kishikawa等人所述,核黄素精确定位在高分辨率冷冻电镜图中,并由紧密的氢键网络结合(扩展数据图6)。我们通过突变NqrB残基D346来确定核黄素的结合位点,该残基与核黄素形成两个氢键。与野生型Na+-NQR相比,D346突变导致电压产生活性大幅下降,生长受损(扩展数据图6)。接下来,我们在冷冻电镜结构中分别以2.09 ?和2.12 ?的分辨率定位了泛素-1和泛素-2。醌类结合在NqrB的边缘,NqrB仍然嵌入在膜中,但靠近Na+-NQR的细胞质方面(图4a,b)。核黄素的醌头基与异alloxazine之间的距离为11.6 ?,适合快速电子转移。
图4:泛醌和抑制剂HQNO在NqrB中具有相同的结合位点。
a,泛素-1 (UQ-1,绿色球体)与NqrB边缘结合,头部基团靠近膜的细胞质面。b,c, UQ-1 (b)和UQ-2 (c)与NqrB的相互作用。类异戊二烯和甲基与残基F159、G141和L138疏水接触,而O4可以与N156的主羰基形成氢键。用其他氨基酸残基取代G141将会在空间上阻断醌类与NqrB的相互作用。NqrB N端的两个两亲性螺旋AH-I和AH-II在UQ结合时关闭,并导致UQ-1或UQ-2与NqrB的主要疏水相互作用。醌头基的甲氧基与AH-II螺旋的L26、A29和L33侧链疏水接触。这些相互作用是至关重要的,并在诱导拟合结合模式下将结合辅因子稳定在该位置。d, Na+-NQR复合物与UQ-1或HQNO的结构比对。两种分子的结合位点重叠,但头基团和尾基团位于结合位点的不同区域。HQNO的喹诺酮头基团与F160和L33侧链相互作用。HQNO的烷基链覆盖了醌头基的空间,并沿着类异戊二烯尾的轨迹运动。像醌一样,HQNO招募n端两亲性螺旋形成高亲和力的结合位点。
抑制剂HQNO结合在相同的结合口袋中,尽管位置略有不同,并阻断醌的进入(图4c和扩展数据图7)。对接计算进一步证实了醌和HQNO的结合姿势(扩展数据图7)。有趣的是,HQNO由铜绿假单胞菌释放7,P. aeruginosa中的Na+-NQR耐受的HQNO水平比霍乱弧菌中的Na+-NQR高16倍8。在序列比对中,我们发现霍乱弧菌NqrB的L33被铜绿假单胞菌NqrB的苯丙氨酸所取代。我们建立了P. aeruginosa的NqrB模型,该模型预测HQNO的结合确实受到苯丙氨酸侧链的阻碍,而醌的结合不受影响。
在cryo-EM和x射线结构中观察到的不同构象在催化循环中被分配给Na+-NQR的不同状态。与UQ-1或UQ-2分离或结合的Na+-NQR的冷冻电镜结构代表了呼吸酶的静止或氧化状态,其中NqrC位于NqrB, NqrF灵活地采用不同的构象,导致该亚基的密度较弱(扩展数据图2)。通过在UQ-2上超过5倍的NADH的冷冻电镜样品获得了还原Na+-NQR的结构信息,这可能导致大多数氧化还原辅助因子的还原。然而,在这种结构中,NqrC位于NqrB,就像处于氧化状态一样。在NqrF中观察到较小的变化,其表现出较少的灵活性,并且在氧化状态下,铁氧化还原蛋白样结构域向NqrF的fnr样结构域移动2 ?。在热力学平衡下,用某种氧化还原态来诱导某种构象似乎是不可能的。引人注目的是,x射线结构显示出与低温电镜结构不同的构象。x射线和低温电镜结构之间最显著的区别是NqrC相对于其他亚基的位置。Na+-NQR的x射线结构显示NqrC插入NqrD-E(图1b和3);然而,在低温电镜测定的所有结构中,NqrC位于NqrB。这有力地说明NqrC在x射线结构中的不同构象是由于Na+-NQR的结晶所致。构象变化不是由于结晶条件的pH值或离子强度,因为这些类似于用于冷冻电镜样品的缓冲条件。我们的结论是,Na+-NQR在晶体中的填充提供了足够的能量来稳定NqrC在NqrD-E的位置。事实上,在x射线结构中观察到的亚基排列在很大程度上是由晶体中分子的堆积而稳定的。NqrC在晶体中与相邻的两个Na+-NQR分子相互作用,形成三个盐桥和几个氢键,从而稳定了代表催化循环某种状态的构象。因为这种状态在热力学平衡状态下没有被观察到,所以它很可能是被晶体填料捕获的一种瞬态或亚稳态。
FMNNqrC与[2Fe-2S]NqrD-E的距离为7.2 ?,表明它代表了跨膜电子转移的快照。用抑制剂DQA处理晶体中的蛋白质的实验表明,这种状态不是很稳定。尽管存在密集的晶体接触,但晶体与抑制剂的孵育引发了NqrC的大构象变化,从NqrD-E向NqrB移动,甚至在晶体内部也是如此。然而,在这种结构中,NqrC并没有达到在低温电镜结构中观察到的NqrB的最终松弛位置,一些残余的晶体触点以瞬态捕获Na+-NQR。
综上所述,我们可以区分出反映不同状态的Na+-NQR的三种构象:(1)在低温电镜结构中观察到的松弛态;(2)在Na+-NQR的x射线结构中表现出短暂的跨膜电子转移态;(3)这两种状态之间的中间状态,是用DQA处理晶体后得到的。
对结构的系统分析表明,不仅NqrC的位置不同,NqrD-E的构象以及NqrC和NqrF的跨膜螺旋的位置和螺旋夹角也不同(扩展数据图8),表明不同亚基的各种运动紧密相连。
考虑到电子从NqrF流向NqrD-E并随后流向NqrC,氧化态的[2Fe-2S]NqrD-E簇必须能够进入NqrF铁氧化还原蛋白样结构域,从而在细胞质侧接受电子。一旦[2Fe-2S]NqrD-E团簇减少,它必须能够从质周侧进入以将电子转移到FMNNqrC。因此,我们提出NqrD-E的不同构象是由[2Fe-2S]NqrD-E簇的氧化还原状态控制的,不同的构象促进了NqrC与NqrD-E在细胞质侧的相互作用或NqrF的铁氧化还原蛋白样结构域与NqrD-E在细胞质侧的相互作用。NqrF铁氧化还原蛋白样结构域或NqrC对[2Fe-2S]NqrD-E簇的交替访问在结构中得到了清晰的记录(扩展数据图8和补充视频1和2)。此外,我们可以将向细胞质侧开放的构象分配给[2Fe-2S]NqrD-E簇的氧化态,将向外质开放的构象分配给[2Fe-2S]NqrD-E簇的还原态。将NqrD- e从细胞质侧的开放构象([2Fe-2S]NqrD- e团簇氧化)转变为周质侧的开放构象([2Fe-2S]NqrD- e团簇还原)的构象变化尤其涉及NqrD和NqrE两个亚基的螺旋iii和螺旋iv(补充视频2)。
NqrE的螺旋- iii和半螺旋- iv倾斜和移动6-10 ?。NqrD和NqrE通过反向拓扑相连,形成紧致二聚体。因此,其中一个亚基的构象变化会影响另一个亚基。事实上,NqrD和NqrE之间是紧密耦合的:NqrE的螺旋- iii和半螺旋- iv的构象变化与NqrD的螺旋- iii和半螺旋- iv的构象变化方向相反。
从观察到的结构中,我们可以得出以下结论:具有氧化的[2Fe-2S]簇的NqrD-E在细胞质方面打开,并且从细胞质可以访问[2Fe-2S]NqrD-E(扩展数据图8a-c),而周质侧由螺旋- iiinqrd和半螺旋- ivnqrd向内运动关闭。反之,如果[2Fe-2S]NqrD-E簇减少,则NqrD-E通过向内移动覆盖簇的螺旋- iiinqre和半螺旋- ivnqre在细胞质方面关闭,而通过向外移动螺旋- iiinqrd和半螺旋- ivnqrd在外周质处进入簇。只有在这种状态下,NqrC才能介导快速电子转移(补充视频2)。在NqrD-E中,这些螺旋的协同运动可能是由指向[2Fe-2S]NqrD-E团簇桥接硫的半螺旋i和半螺旋iv的偶极矩触发的。在FeS簇中,吸收电子后的额外负电荷主要集中在簇的配位和桥接硫原子上9。这种额外的负电荷可能会吸引一个半螺旋的正偶极子,并排斥第二个半螺旋的负偶极子,从而促进螺旋的重新定向。
此外,亚基内的额外相互作用可以稳定这些不同的构象。值得注意的是,具有封闭细胞质侧的NqrD- e构象通过在NqrE中的E95侧链和NqrD中的R71侧链之间形成的盐桥来稳定,而在开放构象中,残基位于13 ?的距离(扩展数据图8d)。如前所述,突变的NqrE-E95消除了Na+-NQR10的Na+泵送活性,表明该构象的稳定至关重要。此外,NqrD-E从氧化的[2Fe-2S]NqrD-E团簇变为还原的[2Fe-2S]NqrD-E团簇的变化导致NqrC和NqrF的跨膜螺旋分别倾斜了12°和7°,并向膜的细胞质方向移动了大约4 ?。这些运动对NqrC的可溶性结构域施加拉力,使其向NqrD-E方向移动,并对NqrF的铁氧化还原蛋白样结构域施加推力,使其远离膜(补充视频1)。
NqrB与尿素转运体11和铵转运体12,13非常相似,这表明它代表Na+-NQR2的Na+转运模块。我们在NqrB的细胞质和质周各观察到两个充满水的半通道。值得注意的是,在低温电镜结构中,NqrC阻断了通往溶剂的质周半通道。人们不能通过低温电子显微镜图的强度来区分水和离子;然而,离子的位置可以通过配位球中相互作用原子的数目和几何形状来确定。
两个位置Na-1和Na-2(图5a,b)被确定为离子结合位点,具有典型碱金属离子的主羰基和水分子的配位球15。图密度、平均键距2.3 ~ 2.5 ?、配位数4 ~ 5与Na+非常吻合。这两个位点Na-1和Na-2都位于靠近质周面。Na-1隐藏在NqrB内部,而Na-2位于NqrB表面,靠近NqrC结合区。Na-2位点可以容纳较大的一价阳离子Rb+和Cs+,而Na-1位点则不能,这表明Na-2也可以结合K+(扩展数据图9)。有趣的是,K+激活Na+- nqr16,17。这种激活可能是由于K+结合到该位点时的结构变化调节了NqrC与NqrB的相互作用。
图5:Na+结合位点和Na+NqrB中的-易位途径。
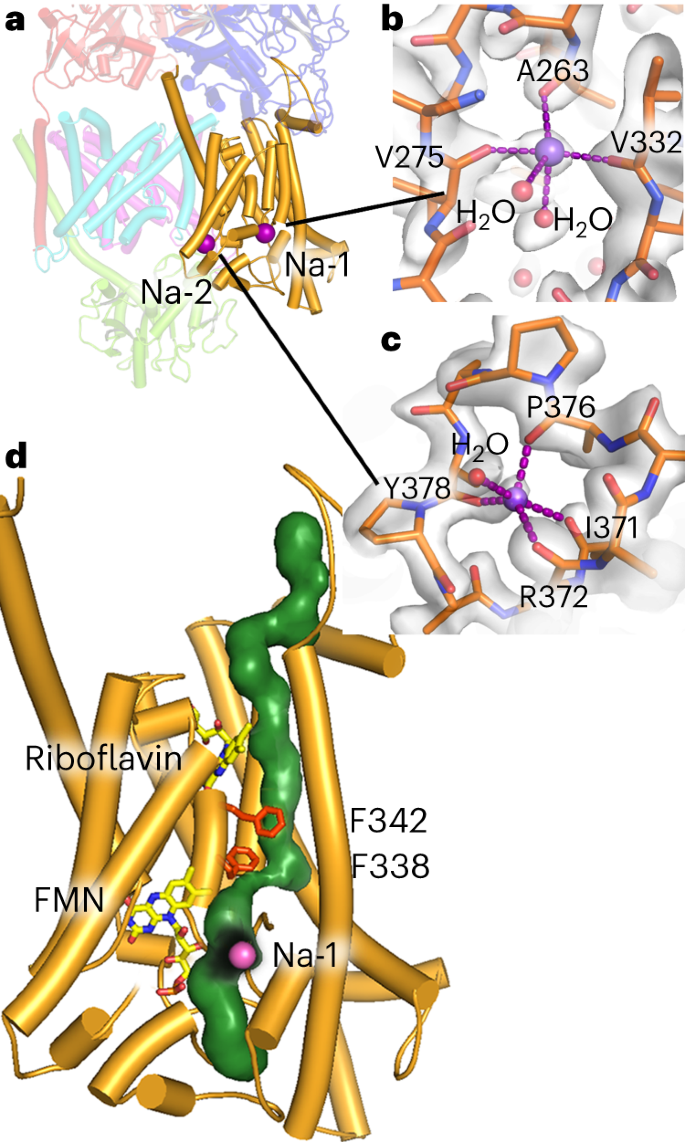
a,在NqrB中鉴定出两个离子结合位点。这两个位点都位于NqrB的质周附近。b, Na-1位点埋在NqrB中,Na+离子由3个主羰基氧原子和2个水分子配位。c, Site Na-2暴露在溶剂中,具有四个主羰基氧和一个水分子的配位球。d,通过NqrB预测Na+易位途径,包括Na-1的位置。易位途径被限制在残基F338和F342附近。
利用HOLE2程序(参考文献18)对高分辨率结构进行分析,证实了NqrB2中存在一个假定的Na+转运途径(图5)。该通路主要由L53、M57、V60、V64、A67、V161、I165、P269、G272、E274、G334和G335等骨干羰基和细胞质入口位置的D52和E157侧链以及质周出口位置的E273侧链组成。这三个酸性残基在不同物种的NqrBs中都是严格保守的。提出的Na+途径还包括Na-1位点,并且在膜的中间被残基F342,特别是F338明显变窄(图5d)。两个苯丙氨酸残基在两个半通道之间形成一个假定的门。有趣的是,F338或F342的改变都会导致Na+依赖性电压形成降低30%,这可能是由于栅极不完全关闭导致易位过程中Na+回流造成的(扩展数据图10)。结果证实NqrB中的F338和F342在Na+易位中起关键作用。
从 

摘要。
主要
结果
讨论
方法
数据可用性
参考文献。
致谢。
作者信息
道德声明
同行评审
扩展数据
补充信息
源数据
这篇文章是由
# # # # #
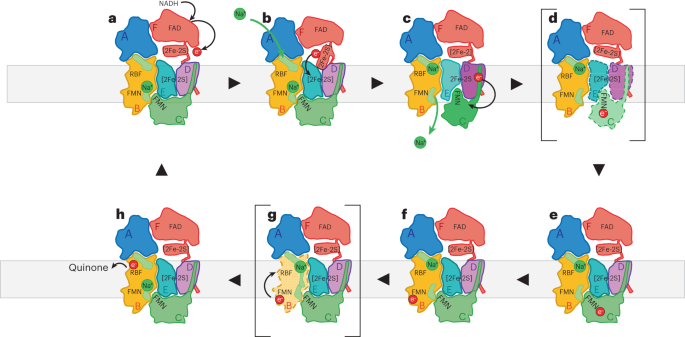
氧化还原驱动离子泵的一个重要特征是电子传递和离子传输之间的严格耦合,以允许对电化学梯度的矢量传输。多中心氧化还原蛋白内典型的电子转移速率在106-107 s?1之间,对应于氧化还原辅因子的边到边距离约为14 ?6。与之形成鲜明对比的是,Na+、K+- atp酶等离子泵的工作速率为50-70 s?1(参考文献19,20)。同样,Na+-NQR的Na+泵送速率为56 s-1(参考文献21)。因此,给定氧化还原辅因子之间的适当距离,电子转移比离子转移快几个数量级。如果所有六个氧化还原辅因子彼此之间的距离都很近,适合快速电子转移,则离子转移过程将太慢,无法连接到几个电子转移步骤;也就是说,不同氧化还原步骤的能量会爆燃。为了将电子转移耦合到Na+泵浦过程中,系统必须在某些步骤上减速电子转移,以允许离子转移进行。这种慢相与Na+-NQR的状态密切相关,在Na+-NQR中,我们观察到氧化还原中心之间的距离很大;也就是说,为了减缓电子转移,氧化还原中心之间的距离必须增加。一旦离子易位发生,构象变化将再次使氧化还原中心在电子转移距离内移动,以实现快速电子转移。
当比较FMNNqrC和[2Fe-2S]NqrD-E在低温电镜结构和x射线结构中的距离时,这种运动是明显的。在cryo-EM结构中,FMNNqrC与[2Fe-2S]NqrD-E的距离为27.3 ?(图1d)。这个距离在x射线结构中缩短为7.2 ?(图1e)。这些距离对应于计算出的大距离的电子转移速率为1.5 × 10?5 s?1,短距离的电子转移速率为1.3 × 107 s?1,其差异系数为~1012(补充表4)。因此,在多步骤过程中,必须有“快速电子转移”和“缓慢构象变化”与“缓慢离子易位”相耦合的交替步骤。“这里提供的数据进一步加强和发展了我们之前在Na+-NQR2中电子转移和Na+易位之间耦合的模型。我们提出了如下文所述的催化循环(图6和补充视频1)。氧化态的Na+-NQR在靠近质周侧的NqrB中有一个Na+结合,这在Na+-NQR的高分辨率低温电镜结构中可以观察到(例如,与UQ-1或UQ-2结合)。一旦NADH与NqrF的fnr样结构域结合,它在FADNqrF处被氧化;在NqrF的类铁氧化还原蛋白结构域中,快速电子转移到[2Fe-2S]NqrF。NqrF的灵活连接的铁氧化还原蛋白样结构域可以接近NqrD-E的细胞质方面,并将还原的[2Fe-2S]NqrF移动到足够靠近膜内的[2Fe-2S]NqrD-E,从而可以转移电子。这一步骤可能会导致NqrB的结构变化,这可能与Na+与NqrB细胞质半通道的结合有关(图6)。膜内[2Fe-2S]NqrD-E簇的减少引发构象变化,在细胞质方面关闭NqrD-E,从而阻止电子回流到[2Fe-2S]NqrF。同时,NqrD-E向外质开放(补充视频2),允许进入NqrC, NqrC从NqrB向NqrD-E移动,使FMNNqrC足够靠近[2Fe-2S]NqrD-E团簇进行电子转移。我们提出NqrC的这种旋转触发了Na+的释放,这些Na+被束缚在靠近质周侧的NqrB中(图6)。一旦FMNNqrC靠近[2Fe-2S]NqrD-E簇,就会发生向FMNNqrC的快速电子转移。NqrD-E与现在氧化的[2Fe-2S]NqrD-E倾向于在质周封闭而向细胞质开放的构象。这促进了NqrC的翻转,减少的FMNNqrC回到NqrB。这种亚基重排可能与胞质半通道中NqrB中Na+的阻断有关。随后从FMNNqrC到FMNNqrB和核黄素inqrb的电子转移步骤可能会触发NqrB细胞质和周质半通道之间的短暂开放。位于NqrB短中心螺旋- viii上的残基B-F338和B-F342在两个半通道之间形成了假定的栅极(图5d)。如先前提出的那样,开放可以通过螺旋- viii和螺旋- x的移位来完成。在相关RNF复合物中的同源亚基RnfD中也提出了类似的螺旋运动22,其中螺旋- x的位置由FMNNqrB的氧化还原状态控制。这种Na+导电状态很可能只是短暂的和瞬态的,到目前为止还没有在任何可用的结构中观察到。在易位时,Na+将驻留在NqrB的质周半通道中,因为出口被NqrC阻断。最后,还原的核黄素inqrb通过醌氧化作为电子受体再循环。
图6:Na的催化循环示意图+-NQR与Na的作用机理+易位。
a、催化循环从NADH在NqrF处被氧化开始。电子被转移到NqrF的灵活系留的铁氧化还原蛋白样域。Na+结合在靠近质周半通道的NqrB中,但离子的释放被NqrC阻断。NqrD-E采用一种允许从细胞质侧进入膜内[2Fe-2S]NqrD-E簇的构象,而在质周侧阻断NqrC的进入。b, NqrF的类铁氧化还原蛋白结构域可以到达[2Fe-2S]NqrD-E簇并转移一个电子。这种电子转移为Na+与NqrB细胞质半通道的结合做了准备。c, [2Fe-2S]NqrD-E的还原触发了NqrD-E亚基的构象开关,现在阻碍了[2Fe-2S]NqrD-E在细胞质侧的进入,但促进了NqrC在质周侧的进入。d,因此,NqrC从其在NqrB的位置向NqrD-E旋转,并触发FMNNqrB附近质周侧Na+结合的释放。NqrC的构象变化使FMNNqrC靠近[2Fe-2S]NqrD-E,导致电子快速向黄素转移。e, [2Fe-2S]NqrD-E团簇的氧化将NqrD-E切换回其先前的构象,触发NqrC向NqrB旋转。f, NqrC与NqrB结合后,电子迅速转移到FMNNqrB。g,我们提出减少的FMNNqrB触发NqrB中两个半通道之间的瞬态打开,使Na+易位到质周半通道。h, NqrB通过电子转移被再氧化为核黄素inqrb,随后被氧化为泛醌。
我们的研究结果提供了人类霍乱弧菌Na+-NQR中电子转移到Na+易位的动态构象耦合的详细模型。这是呼吸复合体如何将氧化还原能转化为化学渗透梯度的例子。Na+-NQR对于大量革兰氏阴性致病菌(包括克雷伯氏菌和假单胞菌等多重耐药菌)的能量节约至关重要,HQNO结合的结构信息为开发新抗生素提供了良好的基础。
Na+-NQR的表达和纯化与前面描述的一样23,并进行了少量修改。简单地说,六组氨酸标记的Na+-NQR在霍乱弧菌Δnqr中表达,而该菌株缺乏NQR复合物的表达。分离膜并用n-十二烷基-β-d-麦芽糖苷(DDM)溶解;Glycon)。Na+-NQR通过Ni-Sepharose Fast Flow (Cytiva)纯化,利用n端六组氨酸标签与亚基NqrA融合。纯化后的Na+-NQR在50 mM磷酸钠、pH 8.0、300 mM NaCl、5%甘油条件下透析。通过超滤(Amicon, 100 kda截止)浓缩Na+-NQR,并使用Superdex 200(16/60)色谱柱(GE Healthcare)在50 mM HEPES-NaOH, pH 8.5, 5%甘油,300 mM NaCl和0.05% DDM中平衡,进一步通过尺寸排除层析纯化。如果需要,在凝胶过滤之前,用携带六组氨酸标签的HRV-3C蛋白酶在冰上进行蛋白水解,从NqrA中去除六组氨酸标签。酶解液中添加0.25 μM的苯基甲基磺酰氟,并再次通过Ni-Sepharose Fast Flow (Cytiva)去除未消化的蛋白质、裂解的六组氨酸标签和HRV-3C蛋白酶。
利用pNqr1、pNB_D346A、pNB_F342A或pNB_F338A等质粒转染缺乏6个nqr结构基因24的霍乱弧菌O395N1Δnqr。按照上述方法纯化Na+-NQR(野生型或变体)。在所有缓冲液中,Na+都被K+取代。缓冲液(50 mM磷酸钾,pH 8.0, 0.05% DDM, 300 mM KCl, 5%甘油)中的Na+-NQR在液体N2中快速冷冻,并在- 80°C保存直至使用。
晶体按照前面描述的方法生长和收集。简单地说,通过超滤(Amicon, 100 kda截止)将Na+-NQR浓缩到7 mg ml -1,并使用Cryschem 24-1 SBS微孔板(Hampton Research)在277 K下通过坐滴气相扩散法进行结晶。将2 μl蛋白溶液与2 μl结晶液(40 mM KSCN, 21.0% PEG 2000 MME, 100 mM Tris-acetate pH 8.5, 8% 1-丙醇)混合形成滴液,在277 K下与100 μl结晶液蒸气扩散平衡。为了进行冷冻保护,将PEG 550 MME加入结晶溶液中,最终浓度分别为0、5、10、15或20% PEG 550 MME,将结晶在每种溶液中浸泡5 - 10秒,并立即在液氮中快速冷冻。为了绘制Na+-NQR中的K+结合位点,晶体在150 mM RbCl或CsCl的存在下生长,数据集分别记录为0.83 ?或1.70 ?。收集了7200度或10000度总振荡的高冗余数据集,以准确确定Rb+和Cs+离子的异常贡献。使用DA+软件26在瑞士光源的X06SA和X06DA光束线收集数据,使用MxCuBE27在同步加速器PETRA III的P14光束线收集数据。所有数据用XDS28进行整合,用XSCALE28进行缩放。使用先前的Na+-NQR (PDB: 4P6V)模型或cryo-EM模型作为搜索模型,用Phaser29进行分子替换来确定结构。异常差图使用阳极(版本2013/1)30,使用来自分子替代的蛋白质模型计算。所有结构均采用Coot(版本0.9.81)31进行迭代模型构建,并使用Refmac5(版本5.8.0350)32和phenix进行细化。精炼(版本1.20.1)在筛选泛素-1、泛素-2、HQNO和核黄素时使用的约束用acedrg(版本246)34从smile代码生成;为了最小化能耗,使用了Refmac5(版本5.8.0350)32。FMN和苏氨酸之间的共价键约束是用acedrg(版本246)生成的。cif文件是使用game(版本18 AUG 2016 (R1))最小化能量后获得的角度和距离手动编辑的。在凤凰提纯过程中使用的谐波约束。Refine33和凤凰。real_space_refine (version 1.20.1)36由phoenix生成[2Fe-2S]簇的配位和几何以及Na+离子的配位。肘部(版本1.20.1)37,如果需要可以手动编辑。
为了制备冷冻电镜样品,将Na+-NQR通过Superdex 200(10/300)柱,在20 mM Tris, 50 mM NaCl, 0.01% DDM, pH 7.6中平衡,并在相同的缓冲液中洗脱。蛋白浓度调整为约4mg ml-1。底物UQ-1和UQ-2分别加入至终浓度1mm。对于含有UQ-2和NADH的样品,底物分别添加至终浓度为1 mM和5 mM。加入HQNO至终浓度为80μM。加入DQA至终浓度为100μM。将3微升样品应用于刚放电的辉光(15 mA,在PELCO easylow系统中持续90秒)C-flat 1.2/1.3 400孔铜网格(Science Services)。样品使用FEI Vitrobot Mark IV在4°C和100%湿度下玻璃化。对于原生Na+-NQR或带有DQA或HQNO的Na+-NQR,在300 kV的Titan Krios G3i (Thermo Fisher Scientific)显微镜上,使用Gatan BioQuantum成像滤光片和K3直接电子检测器,使用EPU软件2.1版(Thermo Fisher Scientific)使用无像差图像偏移(AFIS)自动收集低温电镜数据。视频以×105,000的名义放大倍率获得,其像素大小为0.831 ?。电影被录制为2秒,并被细分为40帧。电子通量率设定为14.5 e -每像素每秒,导致在样品上的累积曝光为41 e - ?-2。使用EPU软件2.1版本(Thermo Fisher Scientific)收集含有UQ-1、UQ-2和NADH + UQ-2的NQR数据,该数据在300 kV的Krios G4上使用冷场发射枪(Thermo Fisher Scientific)和带有Falcon 4探测器(Thermo Fisher Scientific)的Selectris X能量过滤器。采用6-8ev狭缝。NADH + UQ-2数据集的标称放大倍数为×215,000,校准像素大小为0.573 ?, UQ-1或UQ-2的放大倍数为×165,000,像素大小为0.730 ?。数据以猎鹰4号的EER格式收集,总暴露量为~40 e - ?-2。具体请参见表1。
图像处理在RELION4(参考文献)中进行。38、39),除非另有规定。使用RELION实现的MotionCor2算法对剂量分级电影进行运动校正。EER数据以1 e - ?-2的剂量分成几部分。对比传递函数(CTF)在RELION4 (ref. 38)内部使用CTFFIND4.1(参考文献41)进行估计。使用Topaz(0.2.4版本)来挑选颗粒。通过二维分类对数据集进行清理,选取的粒子进行从头重构。在RELION4(参考文献38)中对地图进行了细化,并对数据进行了几轮CTF细化和贝叶斯抛光。作为最后一步,地图处理密度修改使用凤凰。Resolve_cryo_em(1.20.1版本)根据两个独立精炼半图的傅里叶壳相关(FSC) 0.143截断准则估计分辨率39。具体请参见表1。
最初的Na+-NQR模型(PDB: 4P6V)被对接到地图中,并使用Coot(0.9.8.1)进一步构建31。NqrF FAD结构域(PDB: 4U9U)和AlphaFold2(参考文献)的高分辨率晶体结构。44,45)的NqrF氧化还原蛋白结构域模型被用于构建弱NqrF密度。对于nadh结合的Na+-NQR,使用NqrF-F406A突变体的晶体结构。将结构拟合到相应的密度中,并使用LocScale46和phoenix锐化的地图进一步手工构建模型。local_aniso_锐化(版本1.20.1)或phenix。Resolve_cryo_em(1.20.1版本)使用WASP (version 1.0)程序47和CheckMymetal15,48确定假定的离子。
为了进行交联实验,在10 mM HEPES pH 8.0、300 mM NaCl、5%甘油和0.05% DDM的条件下,以1 mg ml-1的浓度制备Na+-NQR配合物。实验用50微升等分液(50μg蛋白质)。Na+-NQR在没有和存在抑制剂DQA的情况下发生交联。DQA从50 mM的DMSO原液中加入到0.5 mM的终浓度。在无抑制剂的样品上加入0.5μl DMSO。交联前,样品在室温下孵育45分钟。
使用同位素轻(d0)和重(d12)二(琥珀酰亚基)亚酸盐(DSS-d0/d12, Creative Molecules)的混合物进行交联。从DMF中制备的25 mM原液中加入终浓度为1 mM的DSS,样品在25°C下孵育45分钟。为了停止反应,从1 M的原液中加入终浓度为50 mM的碳酸氢铵,样品再孵育20分钟。
为了还原和烷基化半胱氨酸残基,首先,将三(2-羧乙基)膦(50 mM水中)加入到终浓度为5 mM,样品在37℃下孵育30 min。随后,加入碘乙酰胺(水中100 mM)至终浓度为10 mM,室温下避光孵育30 min。随后加入0.5μg内源性蛋白酶Lys-C (Wako, 1:100酶底物比),在37℃下消化2.5 h。用50 mM碳酸氢铵(将DDM稀释至CMC以下)将样品溶液稀释至终体积400μl,并加入1μg胰蛋白酶(Promega,酶底比1:50)。蛋白水解在37°C下过夜。
蛋白质消化物采用50 mg SepPak tC18 (Waters)固相萃取纯化,用300μl水/乙腈/甲酸(50/50/0.1,vol/vol/vol)洗脱。蒸发至干燥后,将样品溶解于SEC流动相(水/乙腈/三氟乙酸(70/30/0.1,vol/vol/vol))中,在Superdex Peptide色谱柱(300 mm × 3.2 mm, GE)上,在?kta微型FPLC系统(Cytiva)上,以50μl min-1的流速(参考文献15),通过尺寸排除层析分离肽段。分别收集交联肽优势洗脱范围对应的三个组分进行质谱分析,并蒸发干燥。
干燥的SEC馏分溶解在20μl水/乙腈/甲酸(95/5/0.1,vol/vol/vol)中,4μl在由Easy nLC-1200 HPLC系统和Orbitrap Fusion Lumos质谱仪(均为Thermo Fisher Scientific)组成的液相色谱-质谱系统上进行两份分析。肽在Acclaim PepMap RSLC C18色谱柱(25 cm × 75μm,粒径2μm, Thermo Fisher Scientific)上分离,流速为300 nl min - 1,梯度为89%流动相a /11%流动相B至60% a /40% B, 60 min内分离,其中a为水/乙腈/甲酸(98/2/0.15,vol/vol/vol), B为乙腈/水/甲酸(80/20/0.15,vol/vol/vol)。
以数据依赖的获取模式对肽进行测序,在分辨率为120,000的orbitrap分析仪中获得前体离子扫描。在归一化碰撞能量为35%的线性离子阱中,选择电荷态为+3或更高的最丰富的前驱体进行碰撞诱导解离测序,并以较快的速度在线性离子阱中检测到碎片离子。设置测序周期时间为3秒,一次测序事件30秒后启用动态排除功能。
使用xQuest (version 2.1.4)鉴定交联肽49,50。在Thermo获得的串联质谱数据。原始格式首先转换成。使用msconvert (version 3.0.9134)51对Na+-NQR靶蛋白序列进行检索,并鉴定出污染物(3种人类角蛋白和2种霍乱弧菌蛋白)。DSS特异性设置为仅赖氨酸残基。xQuest搜索的质量误差容限在MS1级别设置为±15 ppm,在MS2级别设置为0.2 Da和0.3 Da(分别适用于普通和xlink离子类型)。
根据实际误差窗口(- 5到+3 ppm或更少,取决于数据集)对搜索结果进行更严格的质量误差容限过滤,并强制执行最小总离子电流值阈值0.1。其余鉴定的光谱都是手工检查的,只保留每个肽至少有四个键切割或每个肽连续三个键切割的鉴定。结合参考文献52的评分方案,xQuest评分阈值分别为18和21.5。对于这种规模的数据库,这通常对应于<5%的假阳性率。
使用xTract(版本1.0.2,可从https://gitlab.ethz.ch/leitner_lab/xtract)49获得)对交叉链接进行量化,并使用默认参数。交联肽对丰度的相对变化以+DQA/ -DQA比值给出。对轻、重DSS产品的电荷状态进行中位数归一化和平均。
配体对接计算使用PLANTS(版本1.2)53、54、SMINA(版本基于AutoDock Vina 1.1.2)55和VINAXB(版本基于AutoDock Vina 1.1.2)56进行。为了与植物对接,利用SPORES57制备了结构。以醌类的密度为中心,以10 ?为半径的搜索范围。为了与VINAXB和SMINA对接,选择了一个16 ? × 18 ? × 16 ?的盒子。在PLANTS (version 1.2)和SMINA或VINAXB中分别在半径为8 ?的球体和12 ? × 16 ? × 12 ?的盒子内对具有短类异二烯尾的UQ-1进行对接计算。结构用凤凰精制。Real_space_refine(版本1.20.1)
从Uniprot (ID: A5F5Y4)中获得了霍乱弧菌NqrF中代表fnr样nadh氧化结构域2的氨基酸序列(129-408残基)。添加一个n端6×His-tag和随后的HRV-3C切割位点,优化密码子在大肠杆菌中的表达。通过GeneArt (Thermo Fisher Scientific)合成编码NqrF129-408和NqrF129-408- f406a的基因,并通过5 ' NcoI和3 ' BamHI分别克隆到载体pET15b中,替换pET15b的6×His-tag和凝血酶裂解位点,生成质粒pFNF53和pFNF406A。
用质粒pFNF53或pFNF406A转化大肠杆菌Tuner (DE3)菌株。细胞在含有100μg ml - 1氨苄西林的DYT培养基中生长,温度37℃。用1 mM IPTG诱导FAD结构域表达,OD600为0.9。30℃下5 h后收集细胞,在10 mM Tris-HCl, 0.3 M NaCl, 5 mM MgCl2, pH 7.4中洗涤。在1 mM DTT和蛋白酶抑制剂(完全不含edta,罗氏诊断公司)存在的情况下,通过约20 kpsi的乳化C-3细胞干扰物(Avestin)一次传代破坏细胞。20000 g离心20分钟,去除细胞碎片。细胞提取液150000 g (Beckman 70Ti型)超离心后,上清过滤后上到Ni Sepharose HP柱(5 ml床体积,Cytiva),用缓冲液a (20 mM Tris-HCl, 0.5 M NaCl, pH 8.0)平衡。用5体积含有30 mM咪唑的缓冲液A洗涤柱,用400 mM咪唑缓冲液A洗脱组氨酸标记蛋白,将各峰组分合并,在50 mM HEPES-NaOH, pH 7.0中稀释至少1:10。用precision蛋白酶在4°C下孵育15 h,将组氨酸标签分离。每1mg蛋白质添加6.7μg蛋白酶。将酶解物上传到Ni+柱上,用50 mM HEPES-NaOH (pH 7.0)洗涤,得到了不含组氨酸标签的FAD结构域。将蛋白上传到SourceQ柱(10 ml床体积,Cytiva)上,用50 mM HEPES-NaOH (pH 7.0)平衡,用0 - 0.4 M NaCl线性梯度洗脱。加入体积5%的甘油,将蛋白质冷冻在液氮中。
结晶前,用ap -5脱盐柱(Cytiva)将缓冲液交换到5 mM Tris-Cl, pH为7.5,蛋白质浓缩到6 mg ml?1 (10-kDa截止Amicon超滤自旋柱,Millipore)。2 μl蛋白溶液与2 μl储层溶液(22-27% PEG 5000 MME, 0.2 M醋酸镁,0.1 M柠檬酸钠,pH 5.0或5.2)混合,在292 K条件下,采用悬滴蒸汽扩散法生长晶体。晶体分别在含有0.2 M NADH (Carl Roth二钠盐)的结晶液中浸泡15 ~ 20 min,或在0.2 M NADH中加入一些二亚硫酸钠颗粒。晶体通过含有35% PEG 5000 MME作为防冻剂的结晶液,并立即在液氮中快速冷冻。使用单色同步辐射(λ=1.0 ?)在X06SA光束线(瑞士光源,Paul Scherrer研究所)采集100 K下的x射线数据。用XDS28程序包对衍射数据进行处理。以NqrF FAD结构域(PDB代码4U9U)为搜索模型,用PHASER29进行分子置换确定结构。在Coot(版本0.9.8.1)31中通过迭代模型构建结构,并使用Refmac5(版本5.8.0350)32和phenix进行细化。精炼(版本1.20.1)
质粒以pNqr1(参考文献23)为模板和特异引物,通过定点诱变构建。点突变通过单次PCR反应(KAPAHiFi PCR Kit, Peqlab)与相应的正向和反向引物导入质粒pNqr1。在Nqr亚基中引入改变的氨基酸残基的突变密码子被下划线。质粒pNF-C70A编码Na+-NQR,携带NqrF中的p.C70A取代。正向引物和反向引物分别为
5 ' - gtatcgttcagcttcagctgcggggggggggggggggggttcatgtg3 '和5 ' - ccacatgaaccaccacccgcagctgaagatacg -3 '。质粒pND-C29A编码Na+-NQR,携带NqrD中的p.C29A取代。正向引物为5′-TTCTGGGTGTGGCGCTGCACTGGC-3′,反向引物为5′-CCAGTGCAGACGCCACACCCAGAAC-3′。质粒pNDE-C29A-C120A编码Na+-NQR,携带NqrD中的p.C29A取代和NqrE中的p.C120A取代。在p.C29A中插入与pND-C29A相同的正向和反向引物。替换p.C120A的正向引物为5′-GATCACAGTAAACGCGGCGATCTTCGG-3′,反向引物为5′-GAAGATCGCCGCGTTTACTGTGATCAGC-3′。质粒pNE-C120S编码Na+-NQR,携带NqrE中的p.C120S取代。正向引物为5′-GATCACAGTAAACTCAGCGATCTTCGGTGG-3′,反向引物为5′-ACCGAAGATCGCTGAGTTTACTGTGATCAGC-3′。
为了获得富含57Fe的Na+-NQR,用pNqr1(参考文献23)或pNF-C70A转化的霍乱弧菌在参考文献2中描述的最小培养基中生长,其中加入1mm MgSO4和1ml SL-9微量元素溶液。SL-9微量元素溶液含有30 mM EDTA, 0.51 mM ZnSO4, 0.62 mM MnCl2 × 4 H2O, 0.097 mM H3BO3, 0.8 mM CoCl2 × 6 H2O, 0.02 mM CuCl2 × 2 H2O, 0.11 mM NiCl2 × 6 H2O和0.16 mM Na2MoO4 × 2 H2O, 0.7 mM CaCl2(参考文献58)。这些化学品纯度最高,铁含量≤0.003%。在这些条件下,霍乱弧菌受到铁的限制,这在添加或不添加56Fe(14μM)的比较生长研究中得到证实。57Fe购自Chemotrade(96.28%同位素富集),为金属粉末,在60℃下溶解于2ml 37% HCl中。用33 ml 0.72 N NaOH中和57Fe原液。培养基和所有溶液均用Millipore H2O配制,通过装有25 g Chelex-100的柱。用0.1 mM EDTA、1 M HCl和chelex处理过的H2O冲洗几次,专门用于细胞生长和纯化富含57Fe的NQR。实验中尽可能地使用了新的塑料器皿。取20毫升经pNqr1或pNF-C70A转化的霍乱弧菌Δnqr24过夜培养物,将1 L新鲜配制的最小培养基与SL-9和14μM 57Fe接种于2 L Erlenmeyer chicane烧瓶(Polycarbonate, Nalgene)中。细胞在37℃下摇培养(180转/分)。在指数后期(OD600, 0.6),添加13mm阿拉伯糖诱导蛋白表达。在25°C (140 rpm)下继续生长20 h。第二天,收集细胞,按照描述23纯化Na+-NQR。通过电感耦合质谱分析证实,Na+-NQR中57Fe的富集量至少为70%。
排除粒径层析后,在NAP-5柱上加入1毫克Na+-NQR进行缓冲交换。蛋白在Tris-HCl pH 8.0, 5%甘油,0.1%(重量)DDM中洗脱,浓缩至约2.5μgμl?1 (Vivaspin 4离心浓缩器,100,000分子量截止,Sartorius Stedim)。用电感耦合等离子体共振质谱法(Spurenanalytisches Laboratorium Dr. Heinrich Baumann)测定Na+-NQR中56Fe、57Fe和S的含量。根据检测到的硫的量,确定样品中Na+-NQR的量。野生型Na+-NQR共含有110种硫(48×蛋氨酸,58×半胱氨酸和2× [2Fe-2S])。为计算S:Na+-NQR比,假设分子量为32.06 g mol?1 (S)和210.8 kDa (Na+-NQR)。为了测定酸不稳定的硫化物,2 nmol Na+-NQR在尺寸排除色谱后进行了三次分析,如文献59所述。
M?ssbauer光谱,镍亲和层析步骤23后,将Na+-NQR浓缩至110-120 mg ml?1至终体积为600μl。将Na+-NQR与66 mM二亚硫酸钠混合。为了使所有辅助因子完全还原,蛋白质在恒定的N2流下与还原剂反应至少5分钟。在400μl M?ssbauer帽中装满蛋白质,并在液氮中进行震荡冷冻。在常规谱仪上记录了γ源交替恒定加速度60的光谱。最小实验线宽为0.24 mm s?1(半高全宽)。温度维持使用M?ssbauer-Spectromag低温恒温器与分裂对磁铁系统(牛津仪器Variox)。γ源(57Co/Rh, 1.8 GBq)室温保存。利用再入式内径管,可以将γ源定位在磁线圈间隙内的零场位置。在300k时,同分异构体的位移是相对于铁金属的。零场下记录的M?ssbauer光谱用洛伦兹双波MFIT程序拟合61。
高能分辨率荧光检测(HERFD)- x射线吸收光谱(XAS)在欧洲同步加速器研究设施(ESRF) ID-26光束线(6 GeV, 200 mA)上进行,配备液氦低温恒温器和20 K的样品室。利用上游的Si(311)双晶单色仪采集铁Kα x射线发射的XAS光谱进行能量选择,校准到能量为7,111.2 eV的铁参考箔的第一个拐点。约翰x射线发射光谱仪配备了四个Ge(440)晶体分析仪,排列在罗兰几何62中。利用能量选择性KETEK探测器,设置在感兴趣的Kα发射荧光区域,进一步提高信噪比。所有EXAFS和XANES光谱都是在Kα最大发射能量(~ 6404 eV)处采集的。评估短XANES扫描(5-60秒)用于评估辐射损伤过程并确定每个点的最大停留时间限制。只有没有显示辐射损伤证据的扫描才被纳入最终分析。样品1 (Na+-NQR含有NqrF中[2Fe-2S]簇,但不含膜内[2Fe-2S]簇)和样品2 (Na+-NQR含有膜内[2Fe-2S]簇,但不含NqrF中[2Fe-2S]簇)的样品斑点总数分别为143和196个(束斑尺寸:1.2 mm (h) × 0.1 mm (v))。原始XAS光谱最初在Matlab(版本R2023a)中平均,并在Athena(版本0.9.26)63中导出进一步处理。在前边缘区域拟合一个二阶多项式,并在整个EXAFS谱中进行相减。使用三区域三次样条(Athena内的AUTOBK函数)对所有光谱的背景函数进行建模,最小k=13.5 1。傅里叶变换在2.0-13.0 1的窗口k范围内进行,所有的傅里叶变换光谱都没有相移校正。
在厌氧室中制备x波段EPR光谱样品。化学药品、溶液和材料在使用前至少保存16小时。缓冲液用N2清洗至少2小时,然后在使用前在厌氧室中储存至少16小时。Na+-NQR (50 mg ml - 1)通过NAP-5柱,用含0.05%(重量)DDM的缺氧缓冲液(10 mM HEPES pH 8.0, 5%甘油,300 mM NaCl)洗脱。收集峰分数,用250μl进行x波段EPR光谱分析。为了实现Na+-NQR的完全还原,蛋白质被允许与二硫代盐(作为粉末添加)反应至少5分钟。连续波(CW) x波段EPR光谱在配备牛津液氮流低温恒温器的布鲁克E500光谱仪上测量,设定温度为10 K,如文献64所述。光谱采集于垂直模式双模x波段谐振器(TE102)中。
在配备1.6 T永磁体的Jasco J-715磁性圆二色谱仪上记录了Na+-NQR (10-20 mg ml?1)的磁性圆二色谱。测量在室温下进行,使用2毫米石英比色管,速度为100 nm min - 1。施加磁场,记录-MCD和+MCD光谱。利用这些光谱计算CD光谱,公式为:+MCD + -MCD=2 CD。累积了三次扫描的数据。
紫外可见光谱在Lambda 12紫外/可见分光光度计(Perkin Elmer)上记录,温度为19°C。凝胶过滤后的Na+-NQR(50-100μM)在10 mM HEPES pH 8.0、5%甘油、300 mM NaCl和0.05% DDM中,在1 mM石英试管中测定吸光度。
利用pNqr1(文献23)作为模板和特异性引物,通过定点诱变技术构建Na+-NQR变体编码质粒,携带NqrB中的替换。点突变通过单次PCR反应(KAPAHiFiTM PCR Kit,来自Peqlab)与相应的正向和反向引物导入质粒pNqr1。质粒pNF-D346A编码Na+-NQR,携带NqrB中的p.D346A取代。正向引物为5′-GTTCTTCATGGCGACTGCGCCAGTTTCTGCGTC-3′,反向引物为5′-GAAGGACGCAGAAACTGGCGCAGTCGCCATGAAG-3′。
质粒pNB_F342A编码Na+-NQR,携带NqrB中的p.F342A取代。正向引物为5′-CGCATTCGGTATGTTCGCTATGGCGACTGACCC-3′,反向引物为5′- gaaactgggtcagtcccatagcgaacataccg -3′。
质粒pNB_F338A编码Na+-NQR,携带NqrB中的p.F338A取代。正向引物为5′- gtatggtggtttcgcagcgggtatgttcttc -3′,反向引物为5′-CCATGAAGAACATACCCGCTGCGAAACCAC-3′。扩增后的载体用DNAse I降解模板载体pNqr1,并在大肠杆菌XL 10-Gold (Stratagene)中转化。为了排除反应过程中其他nqr基因扩增带来的第二位点错误,从大肠杆菌中纯化质粒,用Sal I/Mlu I酶切得到765 bp的片段,每个片段都携带所需的nqrB取代。将这些片段连接到pNqr1的Sal I/Mlu I载体片段(10,568 bp)上,得到载体pNB_D346A、pNB_F342A和pNB_F338A。引入的取代物经测序(Macrogen)证实。对缺乏六个结构nqr基因的霍乱弧菌O395N1 Δnqr的感受态细胞进行了如上所述的转化21。
蛋白脂质体是通过洗涤剂稀释形成的,如前面所述[17,65],但有以下修改。将镍亲和层析纯化的Na+-NQR(野生型或变异型,各2mg / 0.3 mL)在50 mM磷酸钾,pH 8.0, 0.2% DDM, 300 mM KCl和5%甘油中加入脂膜(40 mg l-α-磷脂酰胆碱,来自大豆,II-S型,14-23%为胆碱)中,在圆瓶烧瓶中真空干燥。将脂膜轻轻溶解,加入0.3 ml重构缓冲液(20 mM Tris-H2SO4, pH 8.0, 5%甘油,50 mM K2SO4)。通过滴入0.6 ml重构缓冲液(最终体积1.0-1.2 ml)进一步稀释脂质、洗涤剂和Na+-NQR悬浮液。在1.5 ml的反应管中,用尖端超声器(MS73尖端直径为3mm, Bandelin)对脂质分散和囊泡形成进行了短暂超声检测。尖端放置在蛋白脂质体悬浮液表面以下1cm处。脉冲间隔60 s,脉冲间隔20 s,脉冲振幅30%。为了将洗涤剂浓度降低到临界胶束浓度(CMC)以下,使用滴管(30滴/ min - 1)用50倍体积的重构缓冲液稀释悬浮液。超离心(150000 g, 45 min, 4°C)后,将含有蛋白脂质体的微球悬浮在每1mg Na+-NQR中1ml的重构缓冲液中,对应于每40mg l-α-磷脂酰胆碱中2ml的重构缓冲液中。
NADH: quinoneoxidoreduction
在20 mM Tris-H2SO4, pH 8.0, 50 mM K2SO4, 0.1% DDM (wt/vol)和50 μ M UQ-1条件下(参考文献17),NADH氧化和泛素-1 (UQ-1)被Na+-NQR及其变体(0.4μg蛋白)还原(在氧气存在下)。实验缓冲液中Na+残留浓度为27μM。按规定加入NaCl (0 ~ 30 mM),加入150μM NADH(二钾盐)开始反应。泛醇的形成是亚化学计量的,因为超氧化物是反应的副产物。
停流快动力学
使用配备二极管阵列检测器的SX20止流分光光度计(应用光物理)进行快速动力学测量。在20°C下,在300 nm至1150 nm范围内获得光谱。将缺乏膜内[2Fe-2S]簇的野生型Na+-NQR或Na+-NQR变体(nqrad - c29a)添加到50 mM HEPES pH 8.0, 300 mM NaCl, pH 8.0, 5% (vol/vol)甘油和0.05% (wt/vol) n-十二烷基β-麦糖苷中,并与0.06 mM NADH和1 mM ubiquinin -1混合在同一缓冲液中。反应持续1000毫秒。每次测量重复8次。对得到的光谱进行平均,并使用awk (https://www.gnu.org/software/gawk/gawk.html)提取感兴趣的单个波长。用Origin (OriginLab, https://www.originlab.com/)分析动力学。
从oxonol VI的吸光度差异(630 nm和523 nm)可以看出,蛋白脂质体内部正膜电位的形成。测量前,将蛋白脂质体(3.2 mg脂质中0.16 mg Na+-NQR)与6μM oxonol VI, 50μM UQ-1和NaCl(如所示)混合在实验缓冲液中,该缓冲液与重构缓冲液相同。总检测体积为1.2 ml。10 s后,加入0.2 mM NADH(二钾盐)开始反应,在25℃下搅拌成电压。给出了三个复制的典型轨迹。
构建载体来表达携带半胱氨酸替换的Na+-NQR变体。
将半胱氨酸引入NqrB、NqrC和NqrD亚基的基因片段被合成(Life Technologies),并克隆到pNqr1中(文献23)。质粒pNCD-P174C/Q100C编码Na+-NQR变体,在NqrC中携带p.P174C替换,在NqrD中携带p.p Q100C替换。质粒pNBC-P376C/L226C编码Na+-NQR变体,携带NqrB亚基p.P376C取代和NqrC亚基p.p L226C取代。将质粒转化为缺乏六个结构nqr基因的霍乱弧菌O395N1 Δnqr。
Na+-NQR变体如上文所述被纯化23并在液氮中快速冷冻。在SDS-PAGE69和活性测量之前,等分在冰上解冻并按如下所示处理17。对于SDS - page, Na+-NQR(每道10μg蛋白)与不含还原剂(40 mM Tris-HCl, pH 6.8, 2% SDS, 4%甘油,0.01%溴酚蓝)的上样缓冲液在室温下孵育30分钟。为了促进二硫键的形成,将Na+-NQR变体(每个0.7 mg蛋白)与谷胱甘肽二硫(GSSG)反应缓冲液(229 mM GSSG, 100 mM Tris-NaOH pH 8.0, 200 mM NaCl, 50 mM KCl, 5%甘油,0.05% DDM,总体积为350 μ l)在4°C下孵育48 h。在对照反应中,GSSG被省略。1份同物(20μl)在- 20°C保存,用于后续的SDS-PAGE分析;(另取30μl)保存在4°C,用于随后的活性测定。剩余反应混合物(300μl)上载于nap5凝胶过滤柱(Sephadex G-25, Cytiva),缓冲液(300 mM NaCl, 5%甘油,0.05% DDM, 20 mM Tris-HCl pH 8.0)平衡。Na+-NQR变体用800μl缓冲液洗脱,250μl等分液与50μl 0.8 M dl -二硫苏糖醇(DTT)在0.2 M Tris-HCl中混合,pH 8.0。60 min后,取50μl的反应液用于SDS-PAGE分析,剩余的反应液(250μl)上传到NAP-5凝胶过滤柱(Sephadex G-25, Cytiva)上,用缓冲液平衡。用500μl缓冲液洗脱Na+-NQR变体。在20 mM Tris-H2SO4、pH 8.0、50 mM Na2SO4、0.1% DDM和50μM UQ-1条件下,对分离的、gssg处理的和dtt处理的Na+-NQR变体进行NADH氧化和泛素-1 (UQ-1)还原活性的研究(参考文献17)。
有关研究设计的更多信息可在本文链接的自然组合报告摘要中获得。
ccDownload: /内容/ pdf / 10.1038 / s41594 - 023 - 01099 - 0. - pdf
为您推荐:
- 分析:墨西哥总统竞选明显获胜,但民意调查人员表示投票率至关重要 2025-10-13
- 工商管理学士学位,主办物流会议 2025-10-13
- 哲学最畅销的假日沐浴露现在打四折 2025-10-13
- 新办公室加快马拉维重建 2025-10-13
- 时装周变得政治化:加利亚诺反对法西斯主义 2025-10-13
- Sora是什么?OpenAI新产品超逼真视频惊艳用户 2025-10-13


