双歧杆菌是人类重要的肠道共生菌,特别是在生命第一年的初始微生物群组装期间。双歧杆菌的富集是通过利用人乳寡糖(HMOs)介导的,因为一些适应人类的物种有专门的基因组位点来运输和代谢这些聚糖。这导致发酵产物释放到肠道内,这可能为宿主提供生理上的好处。益生菌物种与同源益生元的共生配对提供了竞争优势,因为益生元提供了营养生态位。
为了确定在2 ' -聚焦乳糖(2 ' - fl)存在或不存在的情况下,hmos分解代谢双歧杆菌菌株的适应性优势和代谢特性,在实验的前3天,常规定植的小鼠被灌胃假atatenulatum双歧杆菌MP80 (B.p. MP80)(作为益生菌)或生理盐水,并在整个研究过程中接受水或含有2 ' - fl的水(作为益生元)。
16S rRNA基因测序显示,在整个实验过程中,只提供b.p. MP80的小鼠与对照组小鼠具有相似的微生物群组成,双歧杆菌科的比例一直很低。1H NMR分析结果显示,对照组和B.p MP80组小鼠肠道内容物和血清代谢谱相似。相反,补充了合成菌的小鼠随着时间的推移,其群落结构发生了戏剧性的变化,口服接种后双歧杆菌科的比例总体增加,但变化不定。根据双歧杆菌科的中位数比例,将合成菌组分为高双歧杆菌持久性和中等双歧杆菌持久性,观察到肠道微生物多样性和代谢物谱的显著差异。值得注意的是,在肠道中双歧杆菌比例高的小鼠中,与2 ' -FL发酵相关的代谢物显著增加,这表明代谢物的产生与种群密度有关。此外,聚焦发酵产物1,2-丙二醇仅在双歧杆菌科高比例小鼠的肝脏和大脑中观察到。
这项研究强调,肠道与共生微生物的定植并不能保证特定的功能输出。
视频摘要
由于益生元的性质,母乳低聚糖(HMOs)的摄入有利于双歧杆菌在母乳喂养的婴儿中早期的显性定植[1]。这些结构复杂的寡糖由一系列单体和键组成[2,3],在肠道中建立营养生态位,选择性地丰富几种双歧杆菌[4,5]。与母乳喂养有关并能进行HMO代谢的分离种包括长芽孢杆菌亚种。婴儿,B.长亚种。长叶芽孢杆菌,短叶芽孢杆菌和伪叶芽孢杆菌[4,5]。在婴儿队列研究中,已经观察到HMO降解、特定双歧杆菌种类的富集、较高的粪便醋酸盐和乳酸盐与有利的健康结果之间的关联[6,7,8,9,10,11]。因此,婴儿期双歧杆菌的强大定植与1型糖尿病标志物的改善[12]、肥胖可能性的降低[13,14]、强大的疫苗应答[15]和较低的抗微生物药物耐药性基因携带[8,16,17]有关。
鉴于长双歧杆菌与婴儿健康结果的关联,人们对促进人类生命后期双歧杆菌种群和该生物的模型定植以仔细研究其作用机制的兴趣越来越大。补充益生菌是一种常用的策略来控制肠道微生物群;然而,功效受到宿主相关因素的个体间差异的影响,包括遗传、饮食和微生物组组成[18,19,20,21,22]。因此,从益生菌中获得健康益处可能是具体情况,只有特定的饮食和/或微生物组配置才能促进代谢或其他有益的微生物活动[23]。虽然益生菌通常能够通过胃肠道存活,但大多数益生菌不会定植,而且它们与本地微生物群和肠道可获得的营养资源的相互作用知之甚少。然而,由益生元组成的协同合成制剂选择性地利用共同施用的益生菌可能会增强肠道中的定植和功能[24]。有了这种有针对性的富集策略,就更有可能通过定殖实现健康结果。
一个物种的持久性被定义为其在一个区域内出现和灭绝之间的时间[25]。营养物质的可用性、环境条件和物种之间的竞争都会影响微生物的生存。细菌持久性由微生物以与冲洗相等或更高的速率复制组成[26]。先前的一项啮齿动物合成模型将发酵乳制品和五种食源性细菌菌株配对,发现在喂养期间,粪便中添加的细菌是可量化的,只有一小部分老鼠继续排出一种细菌,乳酸乳球菌亚种。5个菌株在补充后2天的乳糜泻[27]。研究人员得出的结论是,一小部分老鼠对益生菌耐受,而其他老鼠对益生菌耐受。另外,通过利用已建立的、进化选择的互补乳聚糖-细菌合成对,我们建立了一个持续存在的伪atenulatum双歧杆菌MP80 (b.p. MP80)群体,这是一种母乳喂养的婴儿分离细菌,持续补充HMO 2 ' - focusyllactose (2 ' - fl)[28]。菌株选择基于b.p. MP80在2′-FL上快速生长和代谢的能力[29]。当小鼠遭受化学诱导结肠炎模型时,合成治疗改善了健康结果并减少了炎症,表明协同保护作用。鉴于益生菌补充母乳喂养的婴儿中存在双歧杆菌植入的证据[30,31],该小鼠持久性模型旨在概括hmo在消耗hmo的双歧杆菌定植中的关键作用。
在这里,我们试图研究提供2 ' -FL如何通过b.p. MP80在小鼠肠道中的定植来增加代谢输出。为了了解2 ' -FL和b.p. MP80之间的关联强度,选择了具有显著定植抗性的年轻成年小鼠。我们通过评估含有2 ' -FL和b.p. MP80的合成治疗小鼠的肠道微生物群和代谢谱,与单独补充2 ' -FL或b.p. MP80的小鼠进行比较,来解决这个问题。这使我们能够测量2 ' -FL对维持肠道双歧杆菌种群和相应代谢物谱的影响。确定原生微生物群如何被益生菌或合成菌定植调节以及由此产生的代谢输出对于理解合成菌如何促进健康结果至关重要。
动物按照加州大学戴维斯分校机构动物护理和使用委员会批准的IACUC协议21900进行饲养。雄性C57BL/6J小鼠(5-6周龄,Jackson Labs)分组饲养(每笼3只),保持在22°C, 12 h明暗循环。在开始实验之前,小鼠在该设施中至少适应了1周。食物(5058辐照小白鼠实验室饮食)和水是免费提供的。2′-FL以10% (w/v)的溶液提供在饮用水中。在37°C厌氧条件下,b.p. MP80在de Man, Rogosa和Sharpe培养基(BD Difco微生物,Houston, TX)中生长,并添加0.05% w/v l -半胱氨酸(Sigma-Aldrich, St. Louis, MO)。给予b.p. MP80 (109 cfu/ml PBS)或磷酸盐缓冲生理盐水(100 μl)灌胃3天。在光照周期开始的1小时内,收集单个小鼠的粪便样本。为了验证,在不同的时间点进行了三个实验试验。虽然各试验的实验方案都得到了保留,但不同队列之间的采样天数和最终时间点确实有所不同。在实验1和2中,分别在基线和第2、4、6天和第9天或第10天的最后时间点收集样本。实验3在基线、第4天和第10天采集样品。小鼠通过CO2窒息安乐死。
从粪便样品(30-100 mg)中提取DNA,使用Quick-DNA粪便/土壤微生物迷你准备试剂盒,目录号:D6010 (zimo, Irvine, CA, USA)。按照制造商的说明,提取方案包括使用FastPrep-24仪器(MP Biomedicals, Santa Ana, CA, USA)在25°C下以6.5 m/s的速度进行2分钟的头部跳动步骤。利用条形码PCR引物F515(5′- cacggtcgkcggcgccat -3′)和R806(5′- GGACTACHVGGGTWTCTAAT-3′)[32]扩增16S rRNA基因的V4区,将其修饰为包含一个适配器区域,以便在Illumina MiSeq平台上测序。扩增子经凝胶电泳验证、组合、纯化后送至UC Davis基因组中心进行文库制备和高通量250bp对端测序。原始测序数据在导入QIIME2-2019.10之前被解复用[33]。分裂放大子去噪算法2 (DADA2)用于质量滤波并确定放大子序列变异(asv)[34]。对引物的Reads进行修剪,并截断长度为230 bp的正向和249 bp的反向。读数小于2000的样本从ASV表中删除。使用QIIME和SILVA核糖体RNA基因数据库v138中的na?ve贝叶斯分类器(SILVA 99% 515F/806R)进行分类[35]。样本被细化到3000个序列。原始16s测序数据的NCBI BioProject ID为PRJNA725904。
在R(4.0.2版)中进行微生物群落统计分析[36]。考虑到双歧杆菌科的变异系数较高,如果双歧杆菌科的水平大于双歧杆菌科的中位数比例(> 50.5%),则将双歧杆菌科的水平归类为高富集(HE),如果双歧杆菌科的水平低于中位数比例(< 50.5%),则将双歧杆菌科的水平归类为中度富集(ME)。双歧杆菌科的中位数比例是从最后一个可用的时间点(所有小鼠的最后一天,除了一个测序失败的小鼠,在这种情况下使用第6天)计算的,在一个更大的队列中,用双歧杆菌种(一只手臂是b.p. MP80)和2 ' -FL灌胃的小鼠。这个名称是为了测试不同反应(HE vs. ME)对合成治疗的关联。
采用Shannon指数(vegan::diversity)测定每个粪便样品的α-多样性。采用线性回归分析b.p. MP80 + 2′-FL处理小鼠中HE和ME双歧杆菌组α-多样性的差异。线性混合效应分析(lme4::lmer)[37]包括稳健的三明治方差估计(clubSandwich::vcovCR)[38]和自由度的萨特思韦特校正(clubSandwich::coef_test)。为了调整由于重复测量而缺乏独立性,采用鼠标ID作为随机效应的随机截取模型。选择哪些协变量包括在模型中使用向后逐步消去完成。
为了评估微生物群落重叠,β-多样性采用UniFrac距离(GUniFrac)测量,并使用非度量多维尺度(NMDS)进行可视化(vegan::metaMDS, k=2)[39]。β-多样性统计分析包括检验分散性(vegan::betadisper)、排列多元方差分析(vegan:: adonis2,999个排列)和事后检验(RVAideMemoire::pairwise.perm)。Manova, nperm=" 500 ")[40]。
利用Morisita-Horn不相似性指数来衡量合成制剂治疗小鼠个体内微生物群落演替。计算基线和灌胃后(第4天)之间的数值。相同的群落的Morisita-Horn指数为0,而完全不重叠的群落的Morisita-Horn指数为1。Morisita-Horn稳定性(vegan::vegdist, method=" horn ")的广义线性模型分析(stats::glm, family=二项(link=probit))包括稳健的三明治方差估计和Satterthwaite校正自由度。
利用Songbird进行差异丰度测试,以确定HE和ME分类群的差异以及微生物代谢物相关分类群的差异。Songbird对所选特征之间的对数次变化进行排序[41]。按细菌科聚合asv进行鸣鸟分析。用于鉴定HE和ME类别之间差异分类群的鸣禽分析包括SYN治疗小鼠在最后时间点的样本,并考虑了实验试验差异。对于分类群与代谢物之间的关系,根据先验分析建议,代谢物水平以中位数为基础分为高/低。然后将这些高/低值与鸣禽和最终时间点16S rRNA基因测序数据在家族水平上进行比较,以确定与代谢物水平相关的细菌家族。在某些情况下,样本可能不具有零丰度的分类群,这使得这些样本的对数比无法计算;如果发生这种情况,我们将排除受影响的样品。使用Shapiro-Wilk检验评估差异丰度对数比的正态性,以确定是否应使用学生t检验或Wilcoxon秩和检验。使用Benjamini-Hochberg程序(stats::p.adjust)应用多个比较修正来解释所有分析。
生成了一个分类树来区分HE和ME双歧杆菌分类的小鼠,由于受试者数量少,最小分裂减少(rpart::rpart, minsplit=2)[42]。
称量结肠内容物,加入500 μL等份的冷冻PBS。然后将样品涡旋2分钟,在冰上孵育5分钟,再涡旋2分钟,然后离心(6000 × RCF, 15分钟,4°C)。将上清液转移到新管中,在miVac样品浓缩器中干燥颗粒以测定干重。在额外的离心步骤(14 k RCF, 10分钟,4°C)后,将上清转移到3 kDa过滤器中,再次离心(14 k RCF, 60分钟,4°C),每个样品将207 μL滤液转移到新管中,并与23 μL内标混合,该内标由4.8 mM DSS-d6组成,含有0.2% NaN3(抑制细菌生长),在99.8%的D2O中。在转移到3毫米核磁共振管之前,使用NaOH或HCl将每个样品的pH调整为6.7至6.9之间。血清在冰上解冻,转移到3 kDa过滤器中。4℃,14000 × RCF离心60min后,取207 μL滤液与23 μL 4.8 mM DSS-d6混合。使用NaOH或HCl将每个血清样品的pH调整为6.7至6.9之间。将解冻后的肝脏样品称重,并在MP Bio Lysing matrix D头跳动管(MP Biomedicals, USA)中加入900 μL的冷冻PBS。样品使用FastPrep-24头加热器(MP Biomedicals, USA)以6米/秒的速度匀浆60秒,并重复共2分钟。肝脏匀浆离心10秒,在冰上冷却1分钟,然后再离心15分钟(14 k RCF, 4°C)。将上清转移到新管中,在14 k RCF和4°C下旋转10分钟。然后将上清液转移到分子量为3-kD的过滤器中,在14 k RCF和4°C下离心45分钟。将207 μL滤液转移到新管中,与23 μL 4.8 mM DSS-d6混合,pH调节在6.7 ~ 6.9之间。脑样品解冻,称重,与550 μL的冷冻PBS混合,然后使用MB Bio Lysing matrix D头打管(MP Biomedicals;美国)在6米每秒1分钟,重复2分钟。样本在冰上旋转下来10年代和孵化1分钟之后,离心15分钟在14 k RCF和4°C上层清液被转移到新管和离心10分钟14 k RCF和4°C其次是过滤3 kD分子量通过离心过滤为45分钟14 k RCF和4°C。将207 μL滤液与23 μL 4.8 mM DSS-d6混合在一个新管中,pH调节在6.7 ~ 6.9之间。
1H核磁共振光谱在298 K下使用Bruker Avance 600 MHz核磁共振光谱仪(Bruker BioSpin,德国)上的NOESY 1H预饱和实验(' noesypr1d ')获得。通过以下参数实现数据采集:8次虚拟扫描和32次瞬态扫描,光谱宽度为12 ppm,总采集时间为2.5 s。在松弛延迟(2.5 s)和混合时间(100 ms)期间施加水饱和度。对得到的光谱进行了零填充的傅里叶变换到128 k数据点,并对自由感应衰减(FIDs)进行了指数化变换,对应于0.5 Hz的线展宽。Chenomx NMR Suite v8.4 (Chenomx Inc, Edmonton, Alberta, Canada)用于手动相位和校正基线光谱。使用Chenomx Profiler对每种代谢物进行手动分配和定量。
使用R (v4.0.2)生成所有代谢物统计分析和图形。使用vegan::metaMDS (k=2,距离=“欧几里得”)生成非度量多维缩放(NMDS)图。组间比较采用方差齐性的学生t检验,否则采用Wilcoxon秩和检验。两组以上采用Kruskal-Wallis秩和检验,事后评价采用Dunn检验。方差齐性检验采用Levene检验(car::leveneTest),正态性评估采用Shapiro-Wilk检验,并观察分位数-分位数图残差的偏差。在适当的情况下,采用Benjamini-Hochberg校正对多重比较进行校正。α < 0.05认为差异有统计学意义。
摘要
介绍
方法
结果
讨论
结论
数据和材料的可用性
缩写
参考文献
致谢
作者信息
道德声明
补充信息
搜索
导航
#####
为了测量补充双歧杆菌菌株b.p. MP80后代谢的变化,将39只小鼠分为四组,分别给予b.p. MP80益生菌(n=9;PRO), 2 ' -FL作为益生元(n=9;PRE), b.p. MP80和2 ' -FL (n=12;SYN),或PBS控制(n=9;CON)。PRO组和SYN组益生菌每天灌胃3天,PRE组和SYN组益生菌每天灌胃10%的2′-FL溶液(图1)。本研究共进行了3个试验,样本量如下:试验1,SYN组和CON组n=3;试验2,PRO, PRE, SYN, CON n=3;试验3,CON (n=3), PRO, PRE, SYN (n=6)。
图1
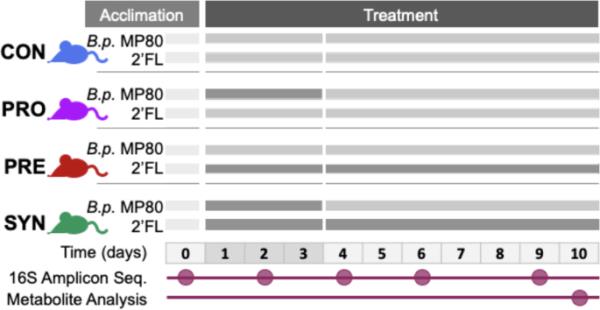
小鼠试验实验设计为时间轴。治疗组分为对照组(CON):口服PBS(1-3天)和饮用水(1-10天),益生菌组(PRO):口服PBS(1-3天)和饮用水(1-10天),益生元组(PRE):口服PBS(1-3天)和饮用水中的2-FL(1-10天),合成菌组(SYN):口服PBS(1-3天)和饮用水中的2-FL(1-10天)。在整个实验过程中收集粪便样本用于16S扩增子测序,在尸检过程中收集代谢物分析样本
为了评估b.p. MP80在小鼠体内的持久性,采用16S rRNA扩增子测序技术评估了粪便微生物群落结构。SYN组小鼠的微生物群落在每个采样日从基线到最终时间点发生变化(图2a, PERMANOVA, p < 0.001)。事后检验显示基线和随后的时间点之间有统计学意义(p < 0.005)。在实验期间,PRO处理小鼠的群落结构发生了较小的、不显著的变化(图2b, PERMANOVA, p=0.181)。尽管差异很大,但SYN组双歧杆菌科的持续存在,实验结束时双歧杆菌科的比例明显高于PRO组(图2c, Wilcoxon秩和检验,p < 0.05)。两两比较加权UniFrac距离发现,除CON组和PRO组外,各组最后一天的微生物群落结构存在统计学差异(表1,PERMANOVA, p < 0.05),说明单独提供b.p. MP80不能影响微生物群落的成员数。然而,基线β多样性在PRE、CON和SYN治疗组(表1,PERMANOVA, p < 0.001)和三个实验试验(补充表1,PERMANOVA, p < 0.001)之间也存在显著差异。
图2
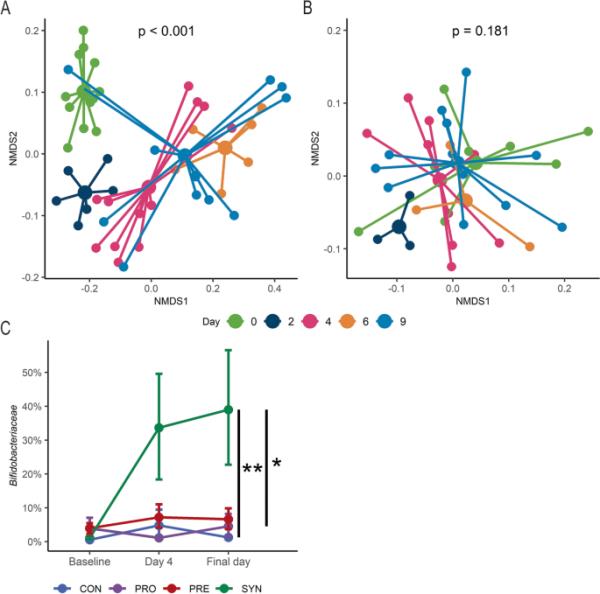
微生物群落结构在合成处理期间的变化。合成小鼠(第0天至最终时间点)微生物群落NMDS图(n=12);B益生菌小鼠(第0天至最终时间点)微生物群落NMDS图(n=9);对照组(n=9)、益生菌组(n=9)、益生元组(n=9)和合成菌组(n=11)从基线、灌胃后(第4天)和灌胃后1周(最后一天)的双歧杆菌相对丰度。最后一天,A组为第9天,根据试验情况分为第9天或第10天。利用β-多样性指数加权二维UniFrac距离生成NMDS。相对丰度数据的误差条表示为自举置信区间的均值和标准误差,迭代1000次。* p < 0.05, ** p < 0.01
表1各治疗组在ba时加权UniFrac指标两两比较的p值线和最终时间点
在完成细菌灌胃后,第4天,我们观察到双歧杆菌科在合成治疗组中的比例变化系数很高(图2c)。因此,我们决定评估这些小鼠体内的微生物多样性,根据接受合成菌的小鼠最终可用时间点双歧杆菌科的中位数相对丰度(50.5%),将组分为高富集(HE)和中等富集(ME)双歧杆菌持久性。α-多样性(Kruskal-Wallis rank-sum检验,p=0.223)和β-多样性(加权UniFrac检验,p=0.256)的微生物群组成在基线时没有显著差异,这表明合成治疗小鼠内部和之间的初始微生物分布相似(补充图1)。我们使用三明治方差的线性混合效应模型,在实验过程中检查了α-多样性,并考虑了基线Shannon指数。当日(灌胃日)是否补充b.p. MP80。有趣的是,与具有ME双歧杆菌持久性的小鼠相比,具有HE双歧杆菌持久性的小鼠α-多样性显著降低(表2)。分类树探索了被分类为HE和ME双歧杆菌持久性的小鼠之间的基线差异。该树鉴定出5只HE双歧杆菌持久性小鼠中有4只具有< 0.9%的丹毒三科相对丰度(图3a)。为了描述HE与ME持续性小鼠肠道微生物群落的稳定性,我们使用广义线性回归和三明治方差来估计个体小鼠从基线到灌胃后一天的森西达-霍恩距离变化,并考虑实验试验。与具有较高b.p. MP80效率持久性的小鼠相比,具有较高b.p. MP80效率持久性的HE双歧杆菌的小鼠具有显着增加的森西塔-角距离,这表明在b.p. MP80灌胃过程中,群落稳定性降低(表3,图3b)。在最后一个时间点,灌胃后1周,HE小鼠的β-多样性与双歧杆菌科的ME持久性相比有显著差异(图3c, PERMANOVA, p < 0.005)。此外,差异丰度测试表明,与ME持久性相比,在HE双歧杆菌持久性分类的小鼠中,双歧杆菌科与毛螺科和瘤胃球菌科的对数比显著更高(图3d, t检验,p < 0.001)。此外,在HE和ME持续组中,Lachnospiraceae的总体中位数比例分别为0.1%和12.4%,而Ruminococcaceae的中位数比例分别低于0.05%和4.1% (Supplementary Fig. 2),进一步反映了合成菌补充小鼠肠道微生物代表的差异。
表2 SYN处理小鼠香农指数(α-多样性)值的线性回归模型
图3
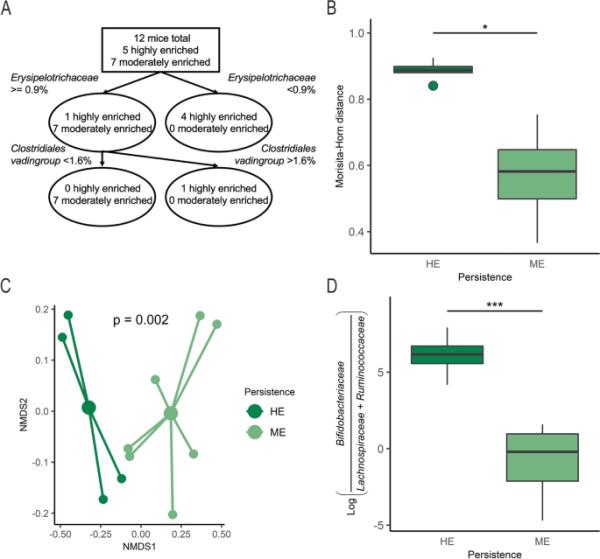
高富集(HE)和中度富集(ME)双歧杆菌分类在合成小鼠体内的微生物群落差异。基于其他微生物类群的分类树来区分HE和ME双歧杆菌持久性小鼠;从基线到灌胃后第一天(第4天),合成治疗小鼠的Morisita-Horn距离分为HE (n=5)和ME (n=7)双歧杆菌;C最终时间点HE (n=4)和ME (n=7)双歧杆菌群微生物群落NMDS图,用高、低标记;试验最后一天双歧杆菌科相对于毛缕菌科和瘤胃球菌科的D对数比。箱形图表示中位数和四分位数范围(IQR),在IQR的1.5倍处,晶须端点等于低于或高于中位数的最大值和最小值。利用β-多样性指数加权二维UniFrac距离生成NMDS。* p < 0.05, *** p < 0.001
表3 SYN处理小鼠的Morisita-Horn (β-多样性)值广义线性回归模型接下来的每一天B.p。MP80填喂法
肠道微生物群的代谢输出本质上取决于微生物的组成、底物的可利用性及其相关的代谢活动。利用质子核磁共振波谱,我们通过在最后时间点取样结肠内容物来询问肠腔的代谢组。非计量多维尺度(NMDS)显示,与CON组小鼠相比,PRO有相当大的重叠,这表明益生菌在补充后1周内不会影响肠道内的微生物代谢物(图4a)。益生元处理的小鼠代谢物谱与其他小鼠不同,主要是由单体聚焦、丙酸和琥珀酸驱动的变化,反映了琥珀酸途径对碳水化合物降解的利用。合成菌治疗导致了高分散,5只小鼠具有不同的肠道代谢物谱,这些代谢物与双歧杆菌集中代谢相关的高乳酸、丙酮酸、甲酸和1,2-丙二醇(1,2- pd)相关。值得注意的是,这些小鼠对应于那些具有HE持久性的小鼠(> 50.5%双歧杆菌科),表明这些代谢物与双歧杆菌定殖程度之间存在关系。利用双歧杆菌科50.5%的截断值,我们比较了NMDS指示的几种有区别的代谢物。乳酸、甲酸和1,2- pd在HE持久性小鼠中均显著升高,而乙酸、丙酸和丁酸在HE持久性小鼠中显著降低(图4b, Wilcoxon秩和检验,p < 0.05)。接下来,我们总结了7种最高浓度的酸,并除以它们的总量,以可视化肠道中有机酸的组成,并通过处理提供相对代谢组成的概述(图4c)。乙酸是主要代谢物,占总有机酸含量的60%以上,SYN组除外,约占总有机酸含量的40%。与NMDS负荷图一致,我们观察到PRE组丙酸盐和琥珀酸盐相对于其他组升高。此外,CON和PRO处理小鼠的有机酸谱相似,与PRE和SYN处理小鼠相比,其丁酸盐比例更高。
图4
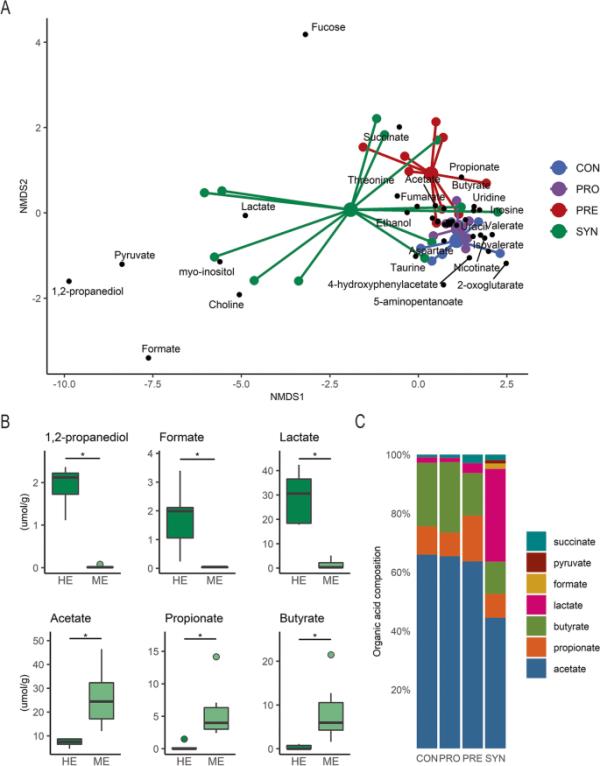
结肠内容物的代谢谱揭示了双歧杆菌高持久性的合成治疗小鼠的不同组成。最后实验时间点小鼠结肠内容物代谢组的NMDS图双歧杆菌科高富集菌(HE)和中等富集菌(ME)在HE (n=4)和ME (n=7)中含量代谢物存在差异;和C有机酸组成。NMDS是利用二维欧氏距离生成的。箱形图表示中位数和四分位数范围(IQR),在IQR的1.5倍处,晶须端点等于低于或高于中位数的最大值和最小值。有机酸的组成是由每种酸的总和除以总量得出的。* p < 0.05
为了进一步评估与益生菌、益生元或合成给药相关的微生物代谢物的变化,对不同治疗组进行了比较。在样品采集的最后一天,CON组和PRO组毛菌科和瘤胃球菌科的比例显著高于对照组(图5a, b, Dunn检验,p < 0.05)。在样品采集的最后一天,PRE处理的小鼠中Bacteroidaceae的比例相对于CON组显著增加,同时自由聚焦浓度更高(图5c, d, Dunn 's检验,p < 0.01),这是之前在Bacteroides消耗HMO时观察到的现象。此外,在提供2 ' -FL (PRE和SYN组)的小鼠中,检测到拟杆菌科与丙酸之间存在很强的相关性(图5e, Pearson ' s r, r=0.741, p < 0.001)。为了评估肠道代谢物与相应微生物群组成之间的关系,我们使用中位代谢物浓度作为截断值来评估总体微生物群落结构的差异和已知产生这些代谢物的选定细菌家族的差异丰度。首先,我们检查了接受2 ' -FL (PRE和SYN组)的小鼠与1,2- pd和丙酸的关系,因为这些代谢物是由病灶分解代谢产生的。在接受2′-FL的小鼠中,双歧杆菌科与毛螺科和Ruminococcaceae的比例显著高于中位数,1,2- pd(一种焦点代谢物)浓度高于中位数(图6a, t检验,p < 0.01)。此外,2 ' -FL喂养小鼠的1,2- pd中位数浓度区分了微生物群落(图6b, PERMANOVA, p < 0.05)。同样,在提供2 ' -FL的小鼠中,高丙酸组观察到拟杆菌科与双歧杆菌科的对数比显著增加(图6c, Wilcoxon秩和检验,p < 0.05),加强了拟杆菌科与高丙酸水平共存之间的联系(图5e)。此外,中位截断点的结肠丙酸盐对微生物群落结构具有歧视性(图6d, PERMANOVA, p < 0.005)。对于所有小鼠,较高的丁酸盐浓度使毛螺杆菌科和瘤胃球菌科的对数比显著高于双歧杆菌科(图6e, Wilcoxon秩和检验,p < 0.01)。然而,由于群体分散不均,在肠道中位数丁酸盐浓度下分裂的微生物群落组成差异无法通过PERMANOVA测试。
图5
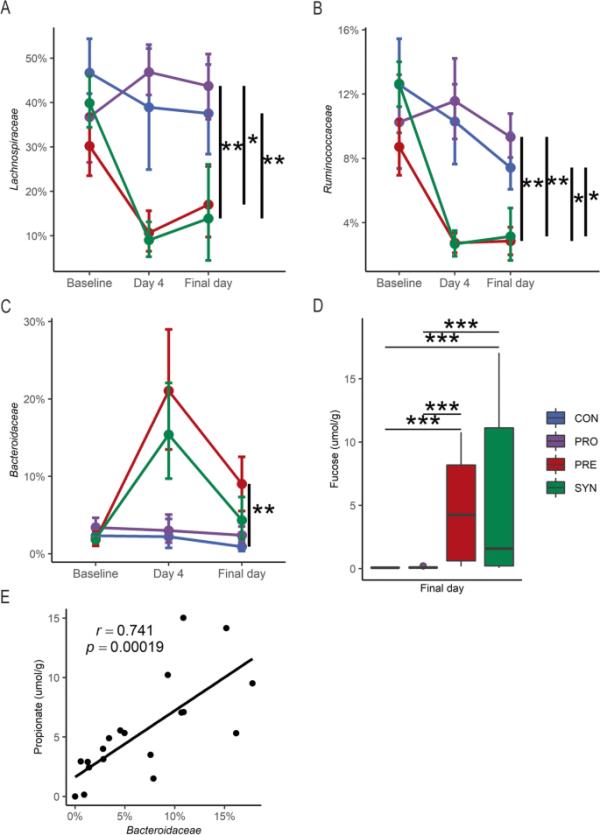
各处理组细菌相对丰度。对照组(n=9)、益生菌组(n=9)、益生元组(n=9)和合成菌组(n=12)从基线、灌胃后(第4天)和灌胃后1周(最后一天)开始的相对丰度,以及所有处理最后一天的游离病灶(μmol/g)的相对丰度。箱形图表示中位数和四分位数范围(IQR),在IQR的1.5倍处,晶须端点等于低于或高于中位数的最大值和最小值。相对丰度数据的误差条表示为自举置信区间的均值和标准误差,迭代1000次。* p < 0.05, ** p < 0.01, *** p < 0.001
图6
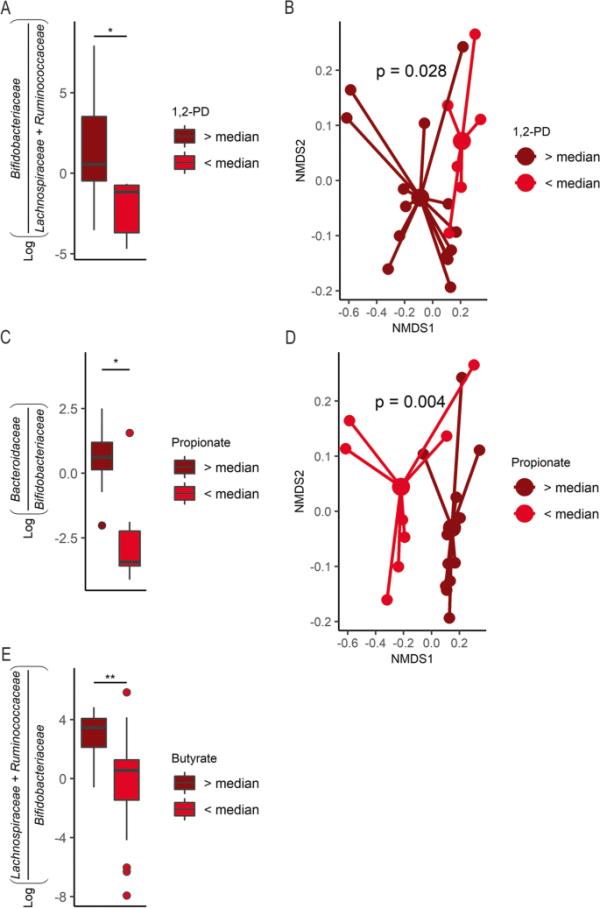
微生物群落的差异与最终时间点不同的代谢物谱有关。所有2 ' -FL小鼠(PRE + SYN)的双歧杆菌科相对于毛螺科和瘤胃球菌科的对数比,分为高于(n=14)或低于(n=6) 1,2-丙二醇(1,2- pd)中位数浓度,B微生物群落NMDS图分为高于和低于1,2- pd中位数浓度;所有2 ' -FL喂养小鼠(PRE + SYN),分为高于(n=12)或低于(n=8)丙酸中位浓度组,拟杆菌科相对于双歧杆菌科的C log比值;D微生物群落NMDS图分组为丙酸中位数浓度以上和以下;E所有处理下毛螺科和瘤胃球菌科相对于双歧杆菌科的对数比,分为高于(n=19)或低于(n=19)丁酸盐中位数浓度。箱形图表示中位数和四分位数范围(IQR),在IQR的1.5倍处,晶须端点等于低于或高于中位数的最大值和最小值。利用β-多样性指数加权二维UniFrac距离生成NMDS。* p < 0.05, ** p < 0.01
最后一天获得的血清进行代谢组学分析,以辨别与治疗相关的全身代谢物的变化。NMDS排序的结果与结肠内容物代谢物相似,CON和PRO组重叠,PRE处理后部分分离,SYN组弥散(图7a)。值得注意的是,与对照组相比,在第一个NMDS轴上表现出最大分离的5只小鼠被鉴定为具有HE持久性(> 50.5%双歧杆菌科)和不同肠道代谢物谱的小鼠(图4a)。血清中与分离相关的代谢特征主要是聚焦、甲酸和1,2- pd。在双歧杆菌持久性高的小鼠结肠内容物中发现的聚焦代谢物1,2- pd浓度较高,在这些动物的血清中也显著较高(图7b, t检验,p < 0.05),表明肠道上皮吸收增加。最后,我们检查了肝脏和大脑,以确定富含微生物代谢物的血液灌注是否与这些器官平衡。值得注意的是,只有经合成菌处理的小鼠在肝脏和脑样品中检测到1,2- pd浓度,而在对照动物中未检测到1,2- pd浓度(图7c)。总之,只有持续定植与循环中观察到的微生物发酵产物1,2- pd的增加相对应。肝脏和大脑中1,2- pd的存在进一步表明在合成治疗过程中代谢物的系统性分布。
图7
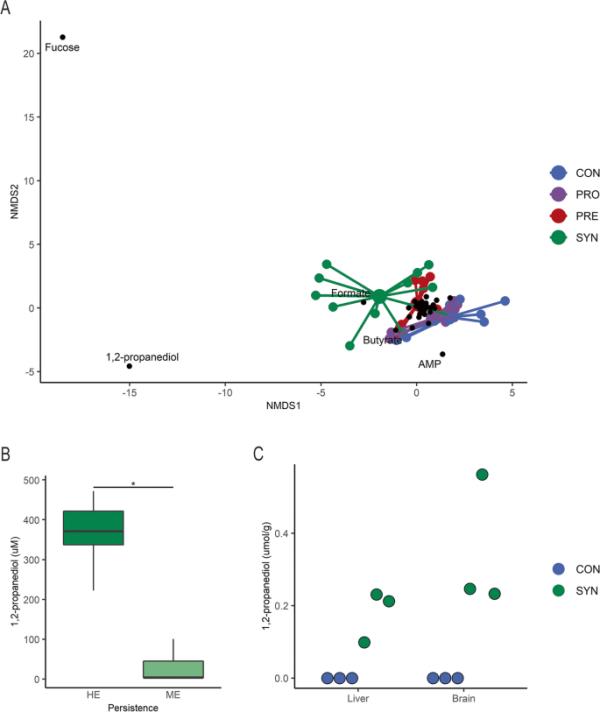
与合成治疗相关的全身代谢组变化。最后实验时间点小鼠血清代谢组的NMDS图,突出显示驱动样品分化的代谢物,以处理组着色;B血清1,2-丙二醇(μmol/g)按高富集(HE)和中度富集(ME)双歧杆菌分类合成处理小鼠;对照(n=3)和合成(n=3)小鼠的肝和脑1,2-丙二醇。NMDS是利用二维欧氏距离生成的。箱形图表示中位数和四分位数范围(IQR),在IQR的1.5倍处,晶须端点等于低于或高于中位数的最大值和最小值。* p < 0.05
在这里,我们证明了b.p. MP80和2 ' -FL在常规定植的小鼠肠道中的合成提供能够调节局部和全身微生物代谢物的输出。双歧杆菌相关的HMO发酵产物在合成菌处理的小鼠中随着b.p. MP80的富集而扩大,而在本研究结束时,仅提供益生菌的小鼠的肠道代谢物谱与未接受益生菌或益生元的小鼠的肠道代谢物谱没有区别。我们之前的研究表明,2 ' -FL足以促进bp MP80在竞争环境中的持久性[28]。与该研究一致的是,我们观察到,尽管在不同的富集水平上,合成菌治疗,而不是单独的益生菌,导致了双歧杆菌的持续定植。在目前的研究中,肠道、肝脏和大脑的代谢物谱提供了一种方法来询问合成治疗对宿主-微生物共代谢的影响。研究表明,个体对入侵微生物的容忍度取决于基线微生物组成[18,19,43],因此,一种新型肠道微生物无法与本地微生物群落竞争已经得到了充分证实。然而,在本研究中,双歧杆菌科HE和ME持久性组之间没有发现基线微生物群落差异(α-多样性或β-多样性)。在整个实验过程中,α-多样性对双歧杆菌高、低比例的分层是明显的,这说明了多样性-入侵效应,即入侵者b.p. MP80的生存与物种丰富度和均匀度呈负相关[44,45]。分类树确定了非常低的基线丹毒科相对丰度与HE双歧杆菌持久性相关。目前,丹毒科的一个未知方面似乎阻止了b.p. MP80有效地利用2 ' -FL营养生态位并击败内源微生物。需要进一步研究丹毒科植物对合生处理的定殖抗性。在最后一个时间点,SYN组内的微生物群落结构(β-多样性)明显不同,这是由于HE和ME双歧杆菌科的持续分裂造成的。
在婴儿中,分解HMOs的功能能力与双歧杆菌及其代谢物的高水平有关[4,17,30,46,47]。在我们的小鼠模型中,一个持续的、占优势的b.p. MP80种群产生了由结肠中乳酸、甲酸和1,2- pd升高所定义的离散代谢谱。此外,这种代谢能力与先前对b.p. MP80 2 ' -FL分解代谢的体外分析一致,使我们得出结论,体内定植的程度与双歧杆菌产生的2 ' -FL代谢物(包括乳酸和1,2- pd)的富集程度相称[29]。由于产生这些代谢物的能力降低和/或它们被其他微生物居民利用,这些产物在成人肠道中的含量相当低[48]。因此,在双歧杆菌高持久性的小鼠中观察到的以双歧杆菌为主的低多样性生态系统更有可能积累这些产物。
由于相关的健康益处,饮食驱动的微生物代谢效应已在人类和动物模型中得到广泛研究[49,50,51]。在这里,我们确定肠道微生物群组成是由它们的代谢输出(丁酸盐、丙酸盐和1,2- pd)来区分的。产生丁酸盐的细菌形成了一个功能群,其中数量最多的两个群体包括直肠真杆菌/Roseburia spp.(毛螺杆菌科)和prausnitzii Faecalibacterium(瘤胃球菌科)[52]。由于丁酸盐的产生是由于到达结肠的复合多糖的分解,因此我们在鼠粮为主要纤维来源的小鼠中发现丁酸盐浓度与毛螺科和瘤胃球菌科的富集有关是合适的。然而,在补充2 ' -FL的小鼠中,丁酸盐浓度成比例地降低,这与先前的一篇报道相吻合,即小鼠补充2 ' -FL与丁酸盐降低有关[53]。在基线之后,我们观察到PRE和SYN组中2 ' -FL的提供与每个采样周期小鼠肠道中拟杆菌科的富集相对应。拟杆菌类通常在其基因组中具有多个多糖利用位点,能够切割包括HMOs在内的多种糖苷键[54]。在代谢方面,拟杆菌是肠道微生物群中通过琥珀酸途径产生丙酸的主要生产者[55,56]。因此,在提供2 ' -FL导致高丙酸的情况下,原生拟杆菌科可能会在益生元喂养小鼠和B.p MP80接受合成的小鼠中竞争本地微生物群落。该模型与先前的报道相似,这些报道描述了母乳喂养婴儿肠道微生物群中拟杆菌的高流行率,这可能是由使用HMOs引起的[57]。拟杆菌和双歧杆菌之间对HMO营养生态位的竞争在该模型中得到了概述[58],仅接受2 ' -FL (PRE)的小鼠中,拟杆菌科的富集程度高于提供双歧杆菌科比例较高的合成菌的小鼠。此外,肠道中含有高浓度1,2- pd的小鼠富含双歧杆菌科。丙二醇途径是双歧杆菌常见的途径,其特点是通过焦点代谢产生1,2- pd,在富含双歧杆菌科的婴儿中发现其代谢水平升高[29,59,60,61]。在我们的模型中,高浓度的1,2- pd仅在双歧杆菌科高的小鼠中观察到,这表明这种代谢物的相关来源。
肠道内产生的一些微生物代谢物可以通过肠道上皮被吸收进入体循环。我们发现每个治疗组表现出相似的血清和结肠内容代谢物谱模式。值得注意的是,在双歧杆菌科的高持久性小鼠中,血清中1,2- pd的浓度明显更高,这表明在该模型中,益生菌的持久性(因此是合成菌的补充)对于产生足够浓度的代谢物以获得全身富集是必要的。这种关系的证据已在人类成人中显示,在此期间提供动物双歧杆菌亚种。乳酸菌(B. lactis)和低聚果糖对血清代谢物的影响比单独使用乳酸菌更明显[62]。此外,微生物代谢产物进入循环的吸收有可能与外周组织相互作用并影响其代谢。除了1,2- pd外,这些组织中的代谢谱在各组之间相似,1,2- pd仅在SYN处理小鼠的肝脏和大脑中检测到。基于这些数据,我们得出结论,肠道微生物发酵产生的高浓度1,2- pd通过门静脉循环经肝脏吸收,随后到达包括大脑在内的其他外周组织。先前使用13C标记的2 ' -FL口服小鼠的研究表明,13C富集发生在肝脏和大脑等组织中[63]。此外,在他们的研究中,给予无菌小鼠的2 ' -FL未能在其组织中保留13C标签,这表明肠道微生物群是2 ' -FL富集的基础。此外,静脉给药导致13C随尿液排出,进一步表明微生物代谢是组织掺入的前兆。我们的工作提供了肠道微生物发酵2 ' -FL产生进入循环的代谢物的证据。这些代谢物很可能在外周组织的代谢中起重要作用,尽管迄今尚未得到证实。考虑到生命早期肠道微生物群的组装可能会调节宿主的代谢过程,从而影响认知和代谢发育,这一点可能很重要。了解微生物代谢物在生命这一阶段的贡献,将有助于开发和使用生物制剂,以赋予整个生命周期的健康。
总之,我们发现将b.p. MP80引入小鼠肠道环境需要同时提供营养生态位(2 ' -FL)来调节局部和全身水平的代谢。如果没有这一优势,当接种益生菌后群落结构重新配置到预处理条件时,定植抗性就无法克服。此外,这强化了hmo作为双歧杆菌竞争性种群的特权营养资源的发现,尽管对定植的许可确实存在可变性。此外,微生物代谢物的富集依赖于b.p. MP80的高度持续居住,并通过宿主体内2 ' -FL分解代谢的双歧杆菌产物反映出来。
我们的研究确实有局限性;虽然小鼠被命令通过相同的设施,但试验之间的基线微生物群落结构不同。这一限制直接影响了asv按细菌家族分组进行数据处理的差异丰度测试。虽然这降低了分析粒度,但细菌科水平上的差异丰度在统计上具有显著性,这表明在本试验中饮食和微生物群落的相关性有多强。此外,各组小鼠的数量在实验试验中分布不均匀,这可能导致基线处理之间的微生物群落差异。小鼠肠道中已建立的微生物群落并不能完美地体现人类肠道微生物群,也不能完全捕捉到对资源和可用物理生态位的竞争。该模型评估了即使在非本地环境中,当提供特殊营养来源时,婴儿携带的双歧杆菌菌株如何具有竞争性,持久性和代谢活性。开发使用合生体的生态框架对于发现与合生体相关的健康结果的潜在机制至关重要。总的来说,这些发现将有助于确定更有可能给宿主带来健康益处的共生配对。
补充表1基线实验试验间加权UniFrac测量的两两比较*。*采用PERMANOVA和FDR校正对比较进行评估。具有统计学意义的p值以粗体表示。补充图1高双歧杆菌和低双歧杆菌治疗小鼠的基线差异不显著。(A)双歧杆菌高富集(HE)组(n=4)和中等富集(ME)组(n=7)小鼠的Shannon α-多样性指数基线值;(B)基线时高、低双歧杆菌群β-多样性指数加权UniFrac的NMDS图。箱形图表示中位数和四分位数范围(IQR),在IQR的1.5倍处,晶须端点等于低于或高于中位数的最大值和最小值。补充图22 ' -FL处理小鼠最终时间点毛螺旋菌科和瘤胃球菌科的相对丰度,根据相对丰度中位数(50.5%)分为高富集(HE) (n=4)和中等富集(ME) (n=7)双歧杆菌科。箱形图表示中位数和四分位数范围(IQR),在IQR的1.5倍处,晶须端点等于低于或高于中位数的最大值和最小值。
下载原文档:https://link.springer.com/content/pdf/10.1186/s40168-023-01624-9.pdf
为您推荐:
- Bayraktar TB3无人机使用滑跃进行首次飞行测试 2025-10-13
- 伍尔弗汉普顿的巨大新仓库现在正在运行,零件公司瞄准“英超联赛” 2025-10-13
- 卡塔尔确保所有居民的健康权:Al Kuwari 2025-10-13
- CE认证简介 2025-10-13
- 报道:高塔姆·辛哈尼亚的离婚大战升温,妻子要求75%的财产 2025-10-13
- 泽连斯基:几名受伤的朝鲜士兵被乌克兰俘虏后死亡 2025-10-13


