提出了一种多残留痕量分析方法,以准确定量146种农药在不同土壤性质的(农业)土壤中。采用快速、简便、廉价、高效、可靠、安全的QuEChERS方法提取农药,采用液相色谱-串联质谱法(三重四极杆)进行化学分析。定量基于基质匹配内标校准,使用95种同位素标记的分析物类似物。与在提取前不久使用新鲜加入分析物的土壤进行方法验证的常见方法不同,我们的方法还通过内部制备的部分老化土壤进行验证,该土壤含有所有目标农药,并通过带有天然农药残留的农田土壤进行验证。该方法灵敏度高(方法定量限中位数为0.2 ng/g),精度高(基于田间土壤,日内和日间方法的中位数精度均为~ 4%),且真实(1)部分老化土壤的农药定量浓度在6个月内保持稳定,接近最初的标称浓度10 ng/g,因此可用于将来的真实性审查;(ii)新加峰相对回收率中位数:103%;(iii)参与环形试验:z分数中位数接近于1(好到令人满意的结果)。将其应用于选定的瑞士(农业)土壤,发现总共存在77种不同的农药,总浓度高达500纳克/克。作为瑞士减少风险和可持续使用植物保护产品行动计划的一部分,该方法现已用于常规土壤监测。

农药用于农田,以对抗或防止害虫、疾病和杂草,以保持作物产量。虽然在过去的十年中,全球农药使用量保持稳定,每年约260万吨[1],但农药对无脊椎动物和植物的毒性却大大增加[2]。这意味着农药对环境的潜在影响不仅取决于消费,而且取决于个别农药对非目标生物的特定高度可变的毒性。尽管欧盟[3]和瑞士[4]针对某些农药制定了基于风险的环境质量标准(EQS)来保护地表水,但在世界范围内,几乎没有针对农药的相关EQS(土壤保护值)来保护土壤生命[5],尽管受农业影响地区的土壤是农药的主要接收方。此外,与来自地表水体的现有监测数据相比,关于农药污染土壤的信息相对较少。然而,最近在瑞士[6,7]和整个欧洲[8,9,10,11,12,13,14,15,16]进行的研究表明,农药在农业土壤中无处不在,甚至在未经处理的地区,如有机管理的田地或广泛管理的草地[17,18]。
为了评估对土壤生命的短期和长期生态毒理学影响,需要进行陆地风险评估。因此,联邦委员会于2017年通过了《瑞士减少风险和可持续利用植物保护产品行动计划》(AP PPP)[19],其总体目标是将农药相关风险降低50%,并推广化学虫害防治的替代品,其中一项措施侧重于发展农业土壤中农药残留的长期监测。这一措施包括地点和物质的选择,开发一种多残留方法来量化土壤中的农药残留,土壤保护值的推导,以及开发农药对土壤肥力影响的生物指标。
土壤是最复杂的环境基质之一,农药与土壤的相互作用通过多种结合过程进行[20,21]。部分施用的农药可以不可逆地与矿物和/或有机物质部分结合,即所谓的不可提取残留物(NER)。NER是否以及在多大程度上永久地结合到土壤基质中,或者不断变化的环境条件是否会导致时间延迟的再动员,目前仍存在争议,且难以测试[22,23]。因此,在农药总库中,区分NER、可分析可提取的总浓度(TEC)和生物可利用浓度是至关重要的,它们使用了不同的概念和定义[24]。绝大多数侧重于监测农业土壤中的农药的研究都依赖于以有机溶剂广泛提取为基础的TEC。
与其他有机污染物(如多环芳烃和多氯联苯)和无机污染物(如重金属)相比[25],据我们所知,不存在老化的认证标准物质(CRM),而这些标准物质会含有大量当前使用的农药。因此,在土壤提取方法的验证过程中,通常的做法是在提取前不久使用加有目标分析物的土壤。然而,基于新添加的土壤样品的回收率预计明显高于那些在老化的土壤。其原因是,由于农药与土壤基质的结合过程复杂且日益强烈,因此分析物与土壤基质的相互作用在陈年土壤和新鲜土壤之间存在很大差异。只有老化的土壤才能反映出与田间土壤相当的提取效率。然而,根据不同的分析方法验证指南,强烈建议使用产生的材料,即目标分析物最初是外来的,但在采样前已被引入的材料(在以下称为含有天然农药的土壤中),并强调了这些材料和加标材料的不同提取效率,但由于缺乏这种CRM,它的使用不是强制性的[26,27]。
2003年推出的QuEChERS[28],一种代表快速、简单、廉价、有效、耐用和安全的提取方法,最初用于从水果和蔬菜中提取农药,已经在很大程度上取代了传统的从土壤中提取农药的方法,如索氏提取、加速溶剂提取(ASE)、超声辅助提取或微波辅助提取[29,30],这些方法耗时且/或溶剂体积要求高。今天,有两种不同的常用缓冲QuEChERS方法,欧洲标准化委员会(CEN)方法EN 15662使用柠檬酸缓冲液[31],AOAC官方方法2007.01使用醋酸缓冲液[32]。
QuEChERS与液相色谱(LC)或气相色谱(GC)耦合串联质谱(MS/MS)相结合已被证明是高效和简单的,具有令人满意的分析性能,最近几项涉及土壤中农药定量的多残留研究已经证明[8,9,10,11,13,15,16,18]。然而,在前面提到的研究中,多个分析方面没有或只是部分解决。这些问题包括:(i)方法验证,在所有研究中,方法验证仅基于提取前不久添加了已知量的分析物的土壤,而没有考虑到老化土壤,即含有天然农药残留的土壤;(ii)部分缺乏敏感性,无法评估亚ng/g范围内的长期生态毒理学影响(方法定量限(MLOQ) 1-20 ng/g [8,9,10,15,16]);(iii)定量依赖于(a)数量有限的同位素标记内标物(ILIS) [9,10,18], (b)根本没有ILIS[8,13,15],或(c)仅将ILIS添加到最终提取物中,而不补偿提取过程中分析物的损失[10]。当使用电喷雾电离(ESI)的LC-MS/MS时,如果定量是基于与分析的现场土壤样品相比具有相似土壤特征(因此在ESI期间具有相似的基质效应)的基质匹配校准,则不使用ILIS的定量是可以接受的。然而,在需要分析许多不同土壤和地点的常规土壤监测中,无论土壤类型如何,ILIS都是必不可少的量化工具。最后,(iv)只有两项研究对土壤中超过100种农药残留进行了定量分析[13,16],而其余的研究在30到80之间。由于欧盟和瑞士批准的植物保护产品中有大量不同的有机合成农药活性成分(200 - 250种[33,34]),而且农药在农业土壤中的存在时间远比其半衰期预测的要长[35],因此应该寻求一种涵盖尽可能多的农药的多残留方法。通过这种方式,可以最全面地评估和描述与农药有关的风险。
本研究采用基于quechers的多残留LC-ESI-MS/MS(三重四极杆)方法,对146种农药(60种杀菌剂、30种除草剂、26种杀虫剂、4种杀螨剂、4种灭鼠剂、2种植物生长调节剂、1种增效剂和19种转化产物)进行了定量分析;根据瑞士植物保护产品条例,SR-916.161[34]在土壤中被列入ESM-A表S2)作为植物保护产品的批准地位。重点放在(i)方法验证,考虑到缺乏原生农药残留的老化CRM, (ii)灵敏度,定量农药残留在亚ng/g范围内,(iii)定量置信和使用~ 100 ILIS来补偿样品制备(提取和进一步的样品处理)过程中潜在的分析物损失,特别是ESI期间土壤特异性基质效应,考虑到需要分析具有不同土壤性质的许多不同土壤。(iv)多残留方法,即对与长期土壤监测相关的大量农药进行量化。它的适用性是通过分析不同的瑞士(农业)土壤的选择。
所有146种分析物和95种ILIS的详细信息(CAS注册号、供应商和纯度)在ESM-A表S1.1中提供。其他化学品和溶剂列在ESM-A表S1.2中。
自制精确浓度(250 ng/mL)的分析物MIX溶液在乙腈中用重量法制备。此外,使用两种外部分析物MIX溶液(从LGC Standards Ltd. (Teddington, UK)和苏黎世州实验室订购[36])进行质量控制。有关不同分析物MIX溶液的详细信息在esm - b1中给出。
同样,在乙腈中制备了精确浓度的自制ILIS混合溶液。根据分析灵敏度调整MIX溶液中各ILIS的浓度;将ILIS分配到三个浓度水平,得到浓度为50-250-750 ng/mL的ILIS混合溶液。通过这种方式,减少了昂贵的ILIS的数量,因为尖峰水平需要适应它们中最不敏感的。
一般来说,所有活性成分(有机和无机)及其主要TPs(如果有符合以下标准的信息)被认为主要在2012年至2019年期间在瑞士被批准为植物保护产品[34](约2700个候选产品)。与长期土壤监测相关的农药(即可能作为残留物存在的农药)然后根据以下三类进行选择:(i)它们的应用(瑞士的使用频率和数量),(ii)它们的环境行为(根据半衰期(DT50)测量土壤退化,根据有机碳-水分配系数(KOC)测量迁移率),以及(iii)它们的生态毒性(根据急性或慢性研究测量毒性,根据辛醇-水分配系数(log KOW)测量生物积累潜力)。如果符合具体标准,并在ESM-B 2中提出了详细的选择程序,则每个类别(i-iii)得分。
基于该方法,145种农药被分类为相关农药和具有LC-ESI-MS/MS多残留能力的农药,其中120种农药被纳入最终分析方法。25种农药被排除在外的主要原因是:(i)它们的疏水性(logKOW高达7),这意味着ESI电离受到阻碍,(ii)快速水解,(iii)没有选择性离子跃迁用于MS检测(三重四极杆),以及(iv)无法获得用于方法开发的参考标准。此外,为了保持过去和未来农药测量的可比性,我们在最终的分析方法中加入了我们实验室[6]的前一种分析方法中的所有农药,尽管其中一些农药(n=23)没有通过选择程序。最后,根据专家意见,又添加了三种农药。因此,在最终的分析方法中总共包含146种农药,其中包括127种母体物质和19种总毒性物质(见ESM-A表S2)。
表1总结了在方法开发(即优化和验证)和应用过程中使用的土壤样品及其主要特征。标准土壤来自LUFA (Landwirtschaftliche untersuchunhe - und Forschungsanstalt Speyer, Deutschland) (S1和S2)和WEPAL, Wageningen University (Wageningen evaluation Programs for Analytical Laboratories, Netherlands) (S3和S4)。土壤S2仅含有六种目标农药的痕量(< MLOQ),主要代表瑞士农业土壤,用于矩阵匹配校准(见“量化”)。
表1用于方法开发和应用的土壤
土壤S2也用于制备标准土壤,其中包含所有目标分析物(S2.1)。用10 mM CaCl2溶液将1千克S2悬浮在300%持水量(WHC)(45.8±2.7 g/100 g)的体积中,并与所有目标分析物加标至10 ng/g干重浓度。这种WHC创造了一个自由流动的土壤-水悬浮液,允许湍流和加标分析物的有效平衡,然后在黑暗中使用TURBULA?摇床混合器(TURBULA;Willy A. Bachofen AG, Muttenz,瑞士)。悬浮液不进行灭菌,以保持土壤结构和土壤特性。相反,选择在5±1°C混合,以尽量减少微生物降解的分析物。下一步,将土壤冻干,再次过筛(≤2mm),并进行湍流。最后,将制备好的土壤保存在琥珀色玻璃瓶中,在- 20°C下避光保存。
制备的参比土S2.1被认为是部分老化的。农药与土壤基质的相互作用时间比新鲜土壤长,因为悬浮、混合和冻干会导致土壤老化;然而,田间土壤样品中的农药可以持续数十年,这取决于它们的土壤耗散,并且与S2.1中的农药相比,肯定会经历更强的结合过程。然而,通过这种方式,所有目标分析物在正确率和日间方法精密度方面的方法验证不仅基于新加标土壤样品的相对回收率,而且还与部分老化的参比土壤S2.1的回收率相辅相成。
此外,在方法开发(S5至S10)和应用(S5至S8和S11至S16)期间,使用了来自瑞士农业现场的本地农药残留土壤样本。此外,来自两个没有农业用途的地点的土壤,即瑞士市政公园(S17)和瑞士国家公园(S18),被列入“阴性对照”。代表性土壤取样(所有表层土壤,0-20 cm)和随后的预处理以及储存条件在esm - b3中进行了描述。
采用QuEChERS从土壤样品中提取农药。所使用的方法基于QuEChERS AOAC方法[32],包括一些修改(在保持样本量与溶剂体积比(1g: 1ml)的情况下,样品的数量、体积和萃取剂的类型,额外的超声和混合步骤,以及省去任何样品清理)。在开始提取之前,使用湍流将干燥和筛分的土壤样品的分布不均性降到最低。取5 g±0.02 g土壤等分称重至50 mL塑料离心管中。下一步,将100μL ILIS MIX溶液(50-250-750 ng/mL)直接加到每个称量的土壤样品上,并使加标ILIS MIX溶液的有机溶剂蒸发至少1小时。然后,在每个土壤样品中加入5 mL纳米水,在短时间内涡旋(~ 10 s),然后使用紊流混合15分钟。将水加入到干燥的土壤样品中,通过使孔隙更容易接近提取溶剂来帮助提取。随后,加入5 mL酸化乙腈(2.5%甲酸(FA)),对样品进行短时涡旋(~ 10 s)。纳米水与乙腈的混合是一个吸热反应;为了释放塑料离心猎鹰管中存在的压力,并避免在混合过程中潜在的泄漏,离心管被短暂地打开并再次关闭。然后用紊流器混合土壤样品15分钟。在下一步中,样本用近10分钟,然后不久涡(~ 10 s),再用10分钟(720 W)。之后,硫酸镁盐混合物组成的4 g, 1 g醋酸钠添加样本立即涡1分钟,混合使用TURBULA 15分钟,然后用10分钟。样本离心机(Rotanta 460 r, Hettich GmbH & Co .公斤,Tuttlingen德国)4分钟在4000 rpm (rcf) 1788。最后,每种上清液1 mL未经过滤转移到LC小瓶中进行化学分析。
土壤S5至S8在未干燥的情况下也进行了分析(见esm - b3)。因此,在取等分之前,使用TURBULA将它们混合,并根据测定的含水量(在16%至25%之间)调整未干土的处理量(5 g±0.02 g),以获得基于干重的相似初始重量。
将开发的QuEChERS方法的提取效率和日内方法精密度等方法性能标准与我们实验室开发的先前应用的略微改进的加速溶剂萃取(ASE)方法进行比较[6](详细信息参见ESM-B 4)。
样品在LC-ESI-MS/MS仪器上测定。使用PAL RTC自动进样器(CTC Analytics AG, Zwingen, Switzerland)将样品注入5μL样品回路(4次过充)。色谱分离使用Kinetex Biphenyl 100 ?柱(100 × 4.6 mm, 5μm粒径,Phenomenex, Torrance, USA),配备C18前柱(4 × 2mm, Phenomenex, Torrance, USA)。流动相由纳米- h2o和甲醇组成,均含有5 mM NH4COOH,液相色谱在35℃下进行,流速为750 μ L/min (Infinity 1290, Agilent Technologies, Palo Alto, USA)。LC运行29 min,优化后的流动相梯度见ESM-A表S3.1。采用ESI进行正负电离,采用三极四极质谱仪(QTrap 5500, Sciex, Toronto, Canada)进行检测,采用预定的多反应监测扫描模式。Q1和Q3(单元隔离模式)的质量分辨率为0.7±0.1 Da。目标周期时间设置为0.6 s,导致每个离子跃迁的停留时间从4.6 ms到167 ms(中位停留时间为13 ms)。
对于每个分析物和ILIS,获得两个离子跃迁,导致总共484个离子跃迁,其中每个分析物和ILIS中较敏感的离子跃迁用作量词,较不敏感的离子跃迁用作限定离子跃迁。所有相关的质谱/质谱参数列于ESM-A表S3.2至S3.5。
土壤S2(标准土LUFA 2.4,土壤特性见表1)制备基质匹配校准曲线。同时对每批(田间)土壤样品提取2次10 g S2,如“土壤提取”所述,提取前不添加ILIS混合溶液,并且使用双倍量的溶剂(纳米- h2o,乙腈(2.5% FA))和盐。将两种S2样品的土壤提取物进行组合和混合。制备浓度为0.05-0.1-0.25-0.5-1-2.5-5-10-17.5-25-35-50 ng/mL(相当于ng/g)的基质匹配校准标准品(每个校准标准品中ILIS的最终浓度为1,5或15 ng/mL;详情见ESM-B 5)。
定量(浓度以ng/g干重表示)基于矩阵匹配的内标校准(MultiQuant定量分析软件3.0.3,Sciex, Canada Toronto),采用线性最小二乘回归,称重因子为1/x。为了构建校准曲线,将每个校准标准品中每种物质的峰面积比(PAR:被分析物的峰面积除以相应ILIS的峰面积)与相应的浓度水平相对应。如果用色谱分离立体异构体(见ESM-A表S3.3和S3.4),则对所有立体异构体的峰面积进行积分和求和。146种分析物中有95种具有结构相同的ILIS (si-ILIS)。对于使用si-ILIS (analytesi-ILIS)的分析物,基于溶剂而不是基质匹配的内标校准将导致同样准确的定量。然而,MLOQ的测定受到阻碍(见“方法验证”),并且忽略了矩阵覆盖对分析物的潜在信号干扰。对于剩余的分析物(analytensi-ILIS),使用非结构相同的ILIS (nsi-ILIS)进行定量,并选择在所有测试土壤(S1至S5)中理想的相对回收率在70%至120%之间的ILIS[27]。为此,使用了相对回收率实验的原始数据(见“方法验证”和ESM-B 9),其中土壤S1至S5分别进行了加标(2.5 ng/g)和未加标的分析。在没有si-ILIS (analytensi-ILIS)的情况下,为每种分析物选择ILIS的系统程序在esm - b6中有详细描述。
计算每个序列中所有矩阵匹配校准标准中每个分析物的平均限定离子与定量离子比(限定离子跃迁峰面积除以定量离子跃迁峰面积)。然后,将同时分析的土壤样品中每种分析物的定性与定量离子比与平均定性与定量离子比进行比较。根据欧盟指令2002/657/EC[38]中定义的标准,允许土壤样品中每种分析物的定性与定量离子比偏离相应的平均定性与定量离子比,并在ESM-B 7中列出。
据我们所知,目前还没有专门针对土壤中农药分析方法验证的指导性文件。因此,遵循SANTE 11312/2021《食品和饲料中农药残留分析的分析质量控制和方法验证程序指南》[27]。与验证相关的术语的定义在不同的指导方针中可能有很大的不同,特别是关于术语准确性、正确性和精度。在本研究中,准确度被定义为ISO 5725-4:2020规定的真实度和精度的结合[39]。
对优化后的QuEChERS提取方法的绝对回收率进行了验证,包括提取的绝对回收率(绝对回收率)、准确度、不同精密度、线性度、选择性、基质效应、MLOQs和仪器定量限(ILOQs)。
绝对回收率根据S1至S3(峰值水平:2.5 ng/g)确定quechers,以确定提取过程中分析物的损失,这些损失会影响所开发方法的性能,例如灵敏度(详细信息参见ESM-B 8)。
通过分析外部参考标准(见“化学品和溶液”)、S2.1的重复分析(6个月内重复分析16种样品制剂)、新添加土壤的相对回收率(S1至S5,使用0.1至10 ng/g之间的不同峰值水平,详细信息参见ESM-B 9)以及参与环形试验(PT-PAS-II:农业土壤中农药的测定,由中央农业监督和测试研究所(úKZúZ),能力测试计划部(OdMPZ)于2022年4月组织。
不同的精密度,即仪器精密度、日内方法精密度(重复性)、日内方法精密度(中间精密度)和人间方法精密度的确定详见esm - b10。
通过将计算浓度与每种校准标准品的加标/实际浓度(允许偏差≤±20%)进行比较,对矩阵匹配校准曲线的线性度进行审查。通过获取每种分析物和ILIS的两个特征离子转移(详细信息请参见ESM-B 7),通过审查限定器与定量器的离子比率(参见“定量”),以及通过将现场土壤样品中每种分析物和ILIS的保留时间(±0.05 min)与同时获得的基质匹配校准标准中的保留时间相匹配来确保选择性。
为了确定土壤特异性基质效应,S1至S5按照“土壤提取”中所述进行提取,但在提取后将ILIS和分析物MIX溶液(lc -瓶中终浓度:2.5 ng/mL)加入土壤提取物中。S1 ~ S5土壤提取物分别用分析物和ILIS混合溶液加标两次和只用ILIS混合溶液加标两次。此外,在乙腈(2.5% FA)中制备了两个浓度为2.5 ng/mL的校准标准品。S1至S5的基质效应计算如下,使用乙腈(2.5% FA)配制的校准标准品中的平均信号强度作为参考(案例(i))或使用S2提取物中的平均信号强度作为参考(案例(ii)):
(1)
其中为每种分析物的平均峰面积,参考案例(i)为,参考案例(ii)为。
这样,矩阵效应用正负百分比表示,正负百分比表示ESI过程中离子增强,负负百分比表示离子抑制。
物质特异性mloq定义为基质匹配的校准标准物的浓度,其中定量剂和限定剂离子的分析物峰面积分别产生至少10和3的信噪比,并且限定剂与定量剂离子的比值符合预期。基于土壤用量为5 g,提取量为5 mL。下一步,通过全局矩阵校正因子调整mloq,该因子根据S1 ~ S5土壤提取物产生的基质效应(有机碳(Corg)含量在1 ~ 5%之间,见表1)扣除。分配该全局矩阵校正因子的原因请参见“定量限制”。通过同时分析几乎无目标农药的S2样品(见“土壤样品”)来考虑空白污染。S2空白样品均未检出目标农药。
通过在纯溶剂(用2.5% FA酸化的乙腈)中注射浓度为0.005、0.01、0.025、0.05、0.1、0.25、0.5、1和2.5 ng/mL的分析物MIX标准物来测定仪器loq,色谱峰必须符合上述相同的S/N标准。
所有图形都是用R 4.2.2版本创建的。[40]。为了说明两组之间的显著差异(例如,通过QuEChERS或ASE获得的个体农药浓度),将其平均值进行t检验(双尾分布,两样本方差相等(均方差),p < 0.05)。事先对两组间的等方差进行f检验(组1和组2的方差无显著差异的双尾概率)。各组间差异百分比计算如下:
(2)
为了比较QuEChERS和ASE得到的农药浓度,除了应用t检验外,还拟合了线性模型。
摘要
介绍
材料与方法
结果与讨论
参考文献
致谢
作者信息
道德声明
补充信息
搜索
导航
#####
质/女士
在ESM-B 11中详细描述了所有离子跃迁选择性、碰撞能量和势以及LC(峰宽)、检测窗口、目标周期时间和停留时间的相互作用的优化和回顾。最终MS/MS获取方法中包含的总共484个离子跃迁信息以及ESI源和气体参数列于ESM-A表S3.2至S3.5。
选择合适的提取方法
与QuEChERS AOAC法相比[32],保持了样品量与溶剂体积比(1 g:1 mL),但将原来的15 g样品量减少到5 g。从样品代表性、灵敏度、线性范围(见“方法验证”)和提取前加入ILIS的必要性来看,这个土壤量被证明是足够的。在提取前加入ILIS的优势体现在相对回收率比绝对回收率中位数高出7%。(在加尖土S1至S3中,总共428个检测中有69%存在显著差异,个体差异列于ESM-A表S4.1;提取前加入ILIS时,S8 ~ S10和S2.1的农药浓度增加,方法验证参数相对回收率和绝对回收率(quechers见“absolute recoveriesQuEChERS”和“Trueness”)。特别是阿特拉辛-2-羟基、异甲草胺ESA、异甲草胺OA和噻苯达唑在提取和进一步的样品制备过程中易发生相当大的损失,在提取前加入ILIS可使农药的定量浓度增加高达80%(个体差异列于ESM-A表S4.1)。
酸性条件被认为可以提高ASE期间土壤中农药的提取效率[7],这就是为什么QuECHERS AOAC[32]方法比CEN[31]方法更受青睐。Acosta-Dacal等人[13]研究发现,使用乙腈(2.5% FA, pH ~ 3.8)萃取剂替代原有的AOAC QuEChERS乙腈萃取剂(1%醋酸,pH ~ 4.2),可进一步提高萃取效率。这一发现在本研究中得到了证实。当用乙腈(2.5% FA)而不是乙腈(1%乙酸)提取S2.1时,单个农药浓度的中位数增加了13%(在总共142次检测中,有86%存在显著差异,个体差异列于ESM-A表S4.2)。相应的,S8至S10的定量个体农药浓度中位数高出19%(在总共99个检测中,有55%存在显著性差异,个体差异列于ESM-A表S4.2)。
通过额外的超声和混合步骤辅助从土壤中提取农药[11,13]。绝对回收率quechers作为提取效率的度量是令人满意的,并在“绝对回收率quechers”中给出。
原始的QuEChERS AOAC方法利用样品清理(通常使用硫酸镁和伯仲胺吸附剂进行分散固相萃取)。当使用LC-ESI-MS/MS时,共提取的基质成分肯定会影响目标分析物的信号强度(见“ESI期间的基质效应”)。然而,已有研究表明,样品清理也会导致回收损失,特别是对于极性更强的农药[13,41]。此外,由于在进样量为5μL时具有优异的仪器灵敏度(使用S2土壤基质的中位基质匹配ILOQ: 0.1 ng/mL),因此不需要清理土壤提取物,使提取方法尽可能简单。
下一步,将优化后的QuEChERS提取方法的提取效率(以获得的浓度衡量)和日内方法精密度与本实验室先前用于定量S2.1和S8至S10土壤中农药的ASE方法[6](详见ESM-B 4)进行比较。两种方法均采用矩阵匹配校准。对于ASE,由于萃取端体积很大,必须在萃取后添加ILIS,因此无法补偿萃取和进一步样品制备过程中潜在的分析物损失。然而,在提取过程中,93%的分析物的绝对回收率在70%到120%之间(个别数据未显示),这表明分析物损失很小,因此可以对所得浓度进行比较,尽管在提取前(QuEChERS)和提取后(ASE)分别添加了ILIS。
使用QuEChERS,在S2.1中获得的单个农药浓度中位数比使用ASE获得的浓度增加了10%(总共142个检测中有49%的显著差异,个体差异列于ESM-A表S4.3),这也反映在线性回归线的斜率上(0.72)(见图1a)。对于S8至S10,线性回归线的斜率接近于1(见图1b),表明QuEChERS和ASE在原生农药残留的农业土壤中的提取效率相当(个体差异列于ESM-A表S4.3)。这些发现与先前关于QuEChERS和ASE提取效率评估的研究[11,42,43]一致。此外,两种提取方法的日内方法精密度没有差异,S2.1和S8至S10的中位数分别为2%和4%(关于QuEChERS方法的日内方法精密度,请参见“精密度”)。
图1
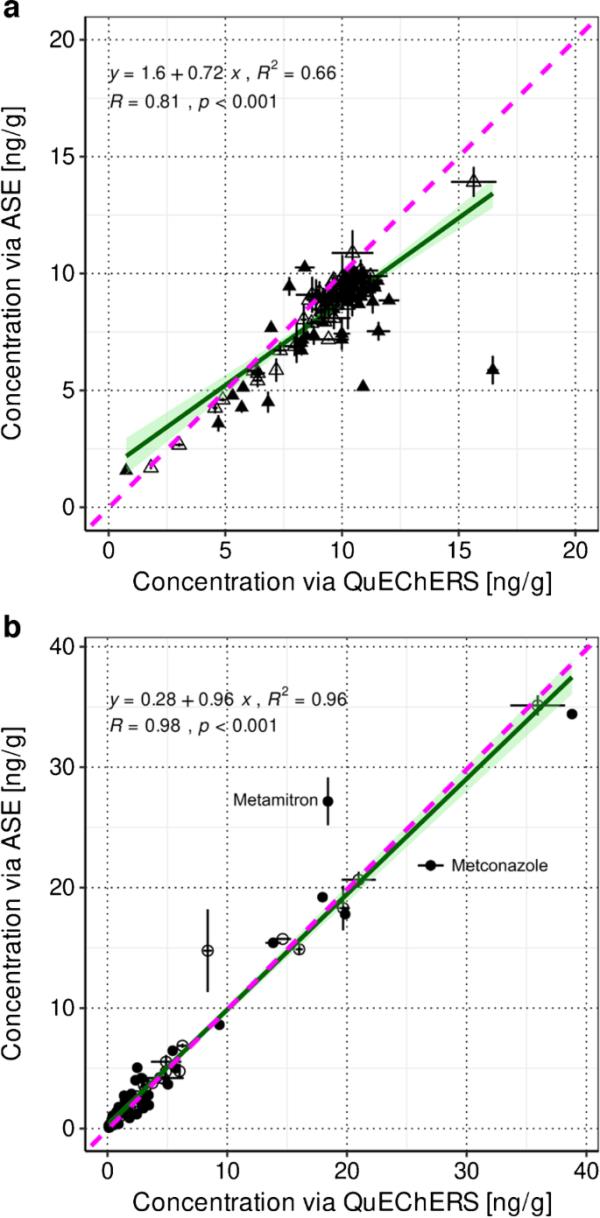
用QuEChERS和ASE提取的土壤S2.1(142个检测,图a)和土壤S8 ~ S10(103个检测,图b)中单个农药平均浓度的1:1线图(两种提取方法的重复数n=2,用ASE提取的S8和S9除外n=3)。当基于t检验(双尾分布,两样本等方差(均方差),p < 0.05)的个体农药浓度与优化后的QuEChERS方法差异不显著时,相应检测结果以开放符号显示。粉红色虚线表示同一性线,绿色线表示具有95%置信区间的线性回归线。此外,还显示了线性方程及其R2和Pearson相关性(R和p值)
然而,当使用多残留方法时,肯定有个别物质用两种方法中的任何一种都表现出更好的提取效率,例如,metamitron (ASE)或metconazole (QuEChERS)(见图1b)。多残留法总是一种折衷的方法,必须根据大多数分析物的结果选择合适的提取方法。
提取前土壤样品处理
为了方便基础设施和子样品的可用性,土壤样品优先存储在环境或稍微冷却的黑暗条件下。然而,这需要通过事先干燥来稳定样品。为了研究土壤样品在中等高温(40°C)下干燥至定重是否会导致农药损失,S5至S8进行了干燥和未干燥分析,即土壤样品在取样后直接冷冻,在筛选< 5 mm之前使用液氮略微解冻,并冷冻保存直到分析(详细信息请参见ESM-B 3)。
干燥和未干燥的S5至S8在数量(在S5至S8中检测到29至37种农药,无论是干燥还是未干燥分析)和检测到的农药的特性以及每种物质的检测浓度范围(两种处理的个体差异和浓度分别列在ESM-A表S4.4和S5中)方面显示出相似的结果。两种方法处理的土壤中测定的总浓度没有差异,并且在4种干燥/未干燥土壤样品中所有检测到的单个农药浓度的日内方法精度(每个处理的4个重复样品制备(干燥或未干燥土壤样品)的中位数相对标准偏差(RSD)内S5至S8: 4%/7%;另见基于干土样品的日内方法精密度“精密度”,两种处理的个别日内方法精密度列于ESM-A表S11.2)。除了分析未干燥土壤样品时的日内方法精度降低外,日内方法精度最高为158%,在所有土壤的129个检测中,有21个检测的日内方法精度高于20%。相比之下,干燥土壤样品的最高回收率为38%,其中6种样品的回收率仅高于20%。这表明干燥的土壤样品比未干燥的土壤样品具有更少的异质性,这允许使用较小的样品等比进行分析。与筛分< 5 mm相比,粉碎筛分< 2 mm明显降低了土壤的非均质性。然而,由于实际原因,冷冻和随后轻微解冻的样品难以处理,并且通过较小的筛孔尺寸在技术上是不可行的。
总的来说,这一比较证明了干燥土壤样品的提取不会面临因温和干燥而失去农药的风险,并指出干燥和未干燥土壤样品的提取效率相似。与未干燥土壤相比,分析干燥土壤的主要优点是更好的日间方法精度,样品制备和储存的实用性以及子样品的可用性,例如,重复分析。虽然农药在储存样品中的长期稳定性在很大程度上仍有待检验,但已证明,至少某些农药的长期稳定性可达8年[35]。
ESI过程中的矩阵效应
基质成分从土壤样品中共同提取。那些最终溶解在萃取物中的物质,如腐植酸,通过LC柱的分析物并与之共洗脱,很可能改变ESI期间分析物的电离效率,从而强烈影响每个被测分析物的灵敏度。因此,研究了Corg含量为~ 1 ~ 5%的S1 ~ S5的基质效应,以反映瑞士表土(0 ~ 20 cm)农田的典型范围[44]。图2显示了以乙腈(2.5% FA)配制的校准标准品中相应的信号强度为参考,所有146种农药在S1至S5土壤提取物中的基质效应分布(案例(i),见“方法验证”)。中位矩阵效应范围为1% (S1)至- 24% (S4)。这些结果证实了基体效应(主要是离子抑制)随着基体复杂性的增加而增加的假设。在ESI过程中,复杂基质成分存在时分析物的离子抑制现象广为人知,并在环境[45,46]、制药[47]、生物分析[48,49]和食品科学[50]等不同领域进行了研究。
图2
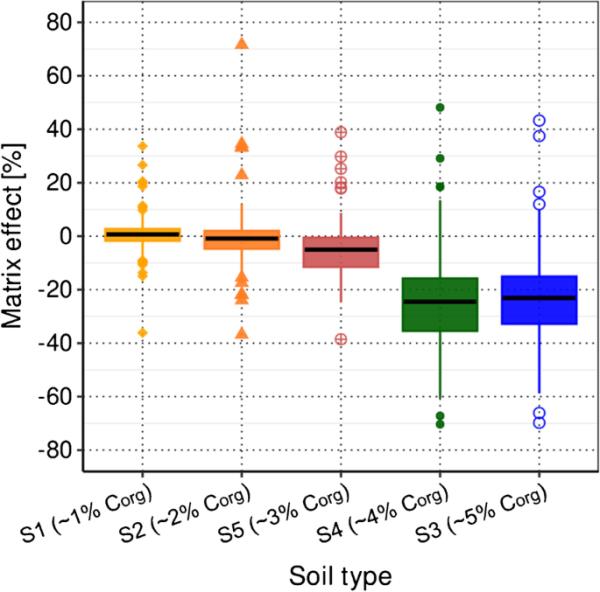
在所开发的分析方法中包括的所有农药在土壤S1至S5中(Corg含量在1至5%之间)的基质效应的箱形图,使用在乙腈(2.5% FA)中制备的校准标准中每种分析物的信号强度作为参考(案例(i));有关详细信息,请参见“方法验证”)
此外,使用S2萃取物中每种分析物的信号强度作为参考,计算S1和S3-S5的矩阵效应(案例(ii);见ESM-B 12图S5和“定量限”)。中位矩阵效应在案例(i)和案例(ii)中没有明显差异。然而,当使用S2提取物(案例(ii))中每种分析物的信号强度作为参考时,最小和最大矩阵效应降低了。使用乙腈(2.5% FA)(案例(i))或S2提取物(案例(ii))中的信号强度作为参考,S1至S5的单个基质效应列于ESM-A表S7中。
他selec分析物无结构相同的ILIS
si-ILIS可用于66%的目标分析物。分析用nsi-ILIS进行定量。然而,选择最适合他analytesnsi-ILIS不是简单,是由系统的评价相对复苏的所有analytesnsi-ILIS——他组合(在一定保留时间窗口,详细描述的标准ESM-B 6)飙升土壤S1的S5 (Corg内容1和5%之间,见表1),最好的结果相对恢复70 - 120%[27]为每个analytesnsi-ILIS土壤基质在每一个测试。
在ESI + (n=40)中电离的分析物-ILIS -ILIS可能组合的中位数为26,在每个分析物的保留时间周围使用±2分钟的保留时间窗口,范围在4到32之间。对于在ESI-中电离的analytesnsi-ILIS (n=11),每个分析物可能有8种analytesnsi-ILIS -ILIS组合,因为在ESI-中电离的ILIS只有8种被纳入分析方法,并且没有施加保留时间限制。
图3显示了S1至S5中分析物-ILIS对分析物-ILIS(无si-ILIS分析物,图A)和分析物-ILIS(有si-ILIS分析物,图B)最终选择的相对回收率。描述的相对回收率用于选择ILIS用于分析的ILIS,基于一个峰值水平(2.5 ng/g),“true”中给出的相对回收率指的是三种不同的峰值水平(0.5,2.5和10 ng/g,见ESM-B 9)。基于应用标准的所有可能的analytessi -ILIS -ILIS组合(ESI +和ESI-)显示在ESM-B 6图S1和S2中。
图3
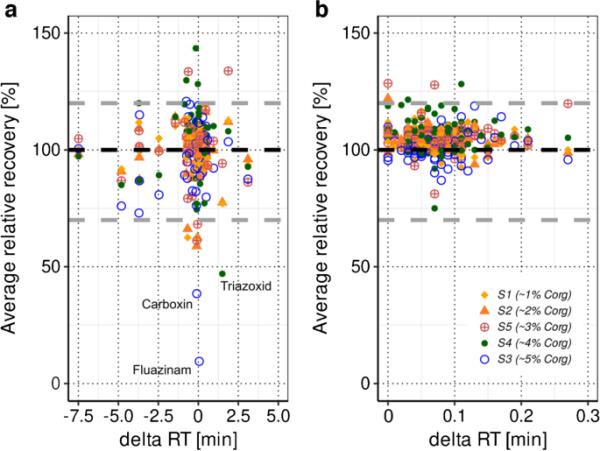
基于最终选择的分析物-同位素标记内标(ILIS)偶对的农药相对回收率,用于分析物-ILIS(结构相同的分析物,图a, n=51个分析物)和分析物-ILIS(结构相同的分析物,图b, n=95个分析物)。显示的是土壤S1至S5的平均相对回收率(土壤S1至S5的四份样品制备)与每种分析物对所选ILIS的保留时间差(δ RT [min])绘制。黑色虚线表示100%的相对回收率,而灰色虚线表示要求的相对回收率范围(70-120%)。
对于analytesnsi-ILIS,所有土壤类型的中位相对回收率为102%,只有6%的analytesnsi-ILIS组合的平均回收率小于70%或高于120%。例如,杀菌剂碳毒素在5种土壤中有4种的相对回收率小于70%。Carboxin易于快速土壤降解(0.5-3.3天[51]),分析方法中包含的ILIS都不能完全弥补这一影响。此外,杀菌剂三氧化二氮和氟唑西南对离子有很强的抑制作用,特别是在高含量的S3和S4提取物中,没有ILIS能够在所有测试的土壤基质中弥补这种作用(见图3a)。
正如预期的那样,对于分析-ILIS, 99%的相对回收率在要求的70-120%范围内(中位数为104%),并且由于ILIS与未标记的分析物几乎相同的物理化学性质,因此分析-ILIS与ILIS之间的中位数保留时间差异为0.07 min。analytesnsi-ILIS和ILIS的中位保留时间差异为- 0.07 min,表明物理化学相似性(保留时间方面)和相同的基质暴露是有效补偿离子抑制/增强的关键。所有分析的最终选择ILIS -ILIS见ESM-A表S6。
最终优化的方法如“方法优化”中所述,通过绝对加样回收率、qechers、正确度、精密度、线性度和定量限(iloq和mloq)进行了验证。选择性(在“方法验证”和ESI - b 7中讨论)和矩阵效应(在“ESI期间的矩阵效应”和“定量限制”中讨论)间接影响这些方法验证参数。
绝对的复苏QuEChERS
考虑到S1到S3, quechers的中位数绝对回收率为95%,而在所有确定的绝对回收率中,只有4%的quechers小于70%(见表2)。这表明在提取和样品制备过程中,分析物损失很小(由于吸附、热降解、挥发或萃取效率不完全)。ESM-A表S8显示了S1至S3中所有农药的单个绝对回收率。
表2总结了土壤中农药痕量分析的最终多残留法中146种农药的优点值
真实
由于缺乏本地农药残留的CRM,在不同的分析复杂性水平上确定了正确度,即(i)基于外部参考标准(见“化学品和溶液”),(ii)部分老化的参考土壤S2.1的重复分析,(iii) S1至S3的相对回收率,以及(iv)通过参与环形试验。
基于S2.1的结果并不总是直接代表真实性。初始加标浓度10 ng/g并不一定对应于恢复的目标浓度,因为有些农药容易形成NER或土壤降解快,如杀菌剂碳毒素(DT50 ~ 0.5-3.3天[51]),其只能恢复~ 0.7 ng/g。然而,根据在6个月内进行的16次独立重复分析,S2.1的中位数浓度为9.3 ng/g(见表2),S2.1中只有27%的单个农药浓度与10 ng/g的峰值浓度偏差超过±20%(单个S2.1浓度列在ESM-A表S9中)。此外,定量农药浓度在6个月内保持不变(日间方法的中位数精度为6%,见“精度”)。因此,从S2.1的这些重复独立分析中获得的平均单个农药浓度可以作为目标浓度,以审查该方法的准确性(在真实度和精密度方面),用于验证和土壤监测常规应用的质量控制。
考虑到所有三种峰值水平(0.5,2.5和10 ng/g),三种新尖刺土壤S1至S3的中位数相对恢复为103%。总体而言,97%的测定相对回收率在70-120%范围内(见表2;个别相对回收率见ESM-A表S10)。如ESM-B 9所述,在开始提取前1小时将分析物MIX溶液加到土壤中,并使有机溶剂蒸发。在初步实验中,分析物MIX加入和提取之间的时间延长至16 h,与暴露时间1 h相比,相对回收率没有差异。
为了外部质量保证,我实验室参与了PT-PAS-II的环形试验。两份来自捷克的农用土壤样品(a、b)中含有本地农药残留,土壤类型为luvisol(由组织者对其代表性进行了测试),以及一份空白土壤材料被运送到24个参与实验室。共有104种农药在环试的目标清单上,其中58种农药被纳入本文的分析方法。z分数的计算基于两种不同的算法,即ISO 13528[52]和Horn程序,其中后者用于参与实验室提交的少量(4-7)每种农药的浓度。测试土壤a和b中88%和93%的检测农药的中位数z-score分别为1.05和1.12(见表2),z-score < 2(标准:0≤|z-score|≤1:良好,1 < |z-score|≤2:满意,2 < |z-score|≤3:可疑),我们开发的分析方法的结果证明是正确的。然而,在29个和23个检测土壤a和b中,分别只有16个和14个检测到与环试验目标农药重叠的农药。其原因是参与实验室提交的农药浓度太少。16种和14种评估农药的所有个体z分数均为正,即本研究中量化的农药浓度始终高于所有提交农药浓度的稳健平均值。由于分析了两种含有天然农药残留的土壤,并且“真实”浓度未知,因此正z分数表明与其他参与实验室相比,提取效率更高。
精度
测定不同的精密度(仪器精密度、日内方法精密度、日内方法精密度和人间方法精密度)以强调方法的性能。总体而言,所有类型的精度都提供了出色的结果,无论是基于新鲜的尖刺土壤,部分老化的土壤S2.1还是农田土壤,精度都具有高度可比性。每种农药的所有精密度列于ESM-A表S11.1至S11.4,表2给出了总结以及为确定所有类型精密度所分析的重复数。
线性
矩阵匹配校准曲线对所有分析物均呈线性关系(R2中值=0.999,R2最小值=0.992),但对单个农药的线性范围不同。线性范围的下限,定义为基质匹配校准标准品的浓度,其中定量剂和限定剂离子过渡的分析物峰面积仍然满足要求的S/N和限定剂与定量剂离子比(见“方法验证”),中位数为0.1 ng/g(最小/最大下限:0.05/5 ng/g),线性范围的中位数上限为35 ng/g(最小/最大上限:5/50 ng/g)(见表2)。每种农药线性范围的下限和上限见ESM-A表S6。
量化极限
中位ILOQ为0.025 ng/mL(见表2;0.125 pg/5μL进样量),所采用的LC-MS /MS系统灵敏度高。个别农药的ILOQs列于ESM-A表s6_。考虑到所开发方法的所有步骤,更相关的是mloq。这些方法应针对具体物质,尽可能敏感,并保持一致,以确保不同土壤和地点之间的数据可比性。然而,由于ESI期间土壤特有的基质效应,这是具有挑战性的(参见“ESI期间的基质效应”)。因此,使用空白土壤基质(标准土壤S2,见表1)进行基质匹配校准,并将其作为确定mloq的起点。空白土壤基质主要代表瑞士农业土壤(标准土壤S2,见表1)。然而,S2只能近似于不同农田土壤提取物中可能发生的基质效应。理想情况下,基质效应必须确定为每个土壤基质报告完全正确的样品特异性mloq。然而,这在常规分析中是不可行的,例如,在长期土壤监测中,为了促进痕量水平的数据评价和统计分析,需要追求一致的最大loq。因此,使用S2中每个分析物的信号强度作为参考(案例ii),确定了S1和S3到S5的矩阵效应(见“方法验证”)。基于这些发现,决定将基于S2的mloq调整为全局基质校正因子2(最大离子抑制为- 50%),因为在开发的方法中,使用S2作为参考的146种农药中,只有三种(阿特拉津-去乙基、阿特拉津-去异丙基和杀虫腈)在任何基质中显示出高于- 50%的离子抑制(见ESM-B 12图S5,个别基质效应见ESM-A表S7)。这一因素相当保守,因为所有土壤中离子抑制的最大中位数为- 24% (S4),根据SANTE 11312/2021指南[27],±20%的基质效应被认为不显著。然而,当在长时间内提取大量不同的土壤时,就像在常规监测中一样,需要寻求可靠的mloq。该分析方法的最小loq中值为0.2 ng/g,约80%的最小loq等于或小于0.5 ng/g(见表2),灵敏度高,适用于土壤监测(个别最大loq见ESM-A表S6)。此外,在低浓度范围内(S2加标0.1和0.25 ng/g)仍然满足方法性能标准,两种浓度水平的相对回收率和日内方法精密度的中位数分别为103%和3%(个别值列于ESM-A表S10)。
与处理土壤中农药分析的其他方法相比,所开发的分析方法的MLOQ灵敏度在一到两个数量级之间(例如,Geissen等[15]的MLOQ(各自研究中包含的农药数量)为1-20 ng/g (36-75);Silva等人[8],10 ng/g (76);?ozowicka等[16],5-10 ng/g (216);kosubov
等[9],3-10 ng/g (64);hv
zdov
等[10],3-10 ng/g (68);Colazzo等[53],1-10 ng/g(30))。只有少数研究报道mloq低于0.5 ng/g (Riedo等[6],0.04-36 ng/g (46);Lafay等[11],0.01-5.5 ng/g (31);Acosta-Dacal等[13],0.5-20 ng/g (218);Homazava等[42],0.1-2.9 ng/g (25);Pose-Juan等[12],0.2-0.7 ng/g (17);Pelosi等[18],0.01-5.5 ng/g(31)),其中大多数mloq低于0.5 ng/g的研究只分析了17 - 46种农药。
总结方法性能
总体而言,所开发的分析方法可对不同土壤性质(土壤pH值为3.6至7.4,Corg含量大多在1至5%之间,见表1)土壤中146种农药进行选择性和敏感的定量分析,其痕量水平主要在亚ng/g范围内。通过使用约100个ILIS及其对analytessi -ILIS的系统分配,确保了准确的定量,以解决分析具有不同土壤特性的许多不同土壤的需求,从而满足土壤特异性基质效应,例如,在长期土壤监测的常规条件下。与仅基于提取前加有农药的土壤样品进行土壤提取方法验证的常见方法相反,我们的方法还通过部分老化的参考土壤S2.1和含有天然农药残留的农田土壤进行验证。这样,我们的真实度和精度等优点(见表2)得到了土壤样品的支持,土壤样品具有更真实的农药与土壤基质的结合亲和力。在常规条件下的质量控制建议在esm - b13中有详细说明。
最后将该方法应用于瑞士常规农业管理农田的8个土壤样品。其中包括四个不同轮作的农田(S5至S8),一个蔬菜地(S11),一个果园(S12)和两个葡萄园(S13和S14)。此外,还分析了两个瑞士草地(S15和S16)、一个瑞士市政公园(S17)和一个瑞士国家公园(S18)的“阴性对照”土壤样本。土壤特性详见表1。
图4显示了所开发方法中146种农药的土壤定量浓度(个别浓度列于ESM-A表S12)。总共有77种不同的农药在所有地点被量化(> MLOQ)。最高的许多不同的杀虫剂被发现农田站点S8和蔬菜站点S11 (n=37),其次是农田网站S6、S7、和S5 (n=32, n=31, n=29日分别),葡萄园S14系列和向(分别为n=21, n=22),果园S12 (n=16),市政公园肌力(n=3),和草原网站S15 S16 (n=3和n=1,分别)。瑞士国家公园(S18)未发现农药,突出了其地处偏远和受保护的地位以及方法的性能,即没有空白污染。与葡萄园或果园等永久文化相比,农田中检测到的不同农药数量较高,这与我们的预期相符。与在永久栽培上的农药施用相比,在农田上的作物轮作导致了更广泛的农药施用模式,在永久栽培上,每个生长季节都施用相同的农药。
图4

选定瑞士(农业)土壤的农药浓度(农田:S5至S8,蔬菜:S11,果园:S12,葡萄园:S13和S14,草地:S15和S16,市政公园:S17,国家公园:S18)。在146个土壤样本中,只有至少一个土壤样本中的农药浓度高于最大限量(MLOQ)。每一行代表一种农药,每一列代表一个地点。农药按农药类别排序,即杀菌剂、除草剂、杀虫剂(包括杀螨剂苯螨酯)和转化产品。在每个农药类别中,农药是根据所有地点的检测频率排序的,然后按字母顺序排序。颜色范围描述浓度水平,空白的白色区域表示浓度低于相应的MLOQ。在8个农业场址(S5至S8和S11至S14)中,最常检测到的三种农药(4-羟基百菌清、2-羟基阿特拉津和二苯醚康唑)以及个体定量浓度最高的农药(氟吡喃)以粗体和斜体突出显示
除草剂检测更频繁地从农田土壤中网站(S5 S8)相比从葡萄园(向和S14系列),在那里~总数的75%检测到农药杀菌剂或TPs(见图4)。这三个最频繁检测农药八农业领域的网站(S5 S8和S11 S14系列)是TP chlorothalonil-4-hydroxy网站(8/8)其次是TP atrazine-2-hydroxy网站(7/8)和杀菌剂difenoconazole(7/8网站)。百菌清是一种广谱杀菌剂,由于其致癌特性和在地下水中检测到潜在毒性的百菌清TPs,于2019年在欧盟和瑞士被禁止使用[54,55]。分析的农业地点是在2016年至2019年初之间采样的,这是在瑞士全面禁止使用百菌清之前。在所有农业站点均检测到的酚类TP -4-羟基表现出中低迁移率(KfOC: 250-718 L/kg[56]),而在4/8个站点检测到的第二种酚类TP R611968 (KfOC: 51-128 L/kg[56])的迁移率增加。相比之下,磺酸TP R417888的流动性更强(KfOC: 5-17 L/kg[56]),仅在1/8的位点发现。这与地下水样品中更频繁检测到的磺酸型百菌清TPs(如R417888)与酚型百菌清TPs(如R611968)相一致,后者流动性较小,因此保留在土壤中[57,58,59]。然而,在上述研究中并没有分析氯他隆-4-羟基。阿特拉津-2-羟基是除草剂阿特拉津的一种总磷,该除草剂已于2005年在欧盟被禁止使用[60],2009年在瑞士被禁止使用[34],在一些研究中已经观察到[6,7,9,17,61],这表明过去阿特拉津的高施用量是遗留问题。在AP PPP中[19],杀菌剂异丙康唑因其持久性(田间土壤降解,DT50: 20-265 d[51])和毒性(无效应浓度(NOEC)蚯蚓:200 ng/g, NOEC大水蚤:0.0056 mg/L[51])被列为高危暴露农药。它在农业土壤中频繁出现,浓度接近陆地noec,这加强了将该农药列为替代候选农药的决定。
在分析的土壤中,杀菌剂及其TPs达到了最高的单个农药浓度(见图4)。特别是在蔬菜场地(S11), flupyram量化了最高的单个农药浓度,最高可达140 ng/g。然而,农业土壤中单个农药浓度在很大程度上取决于采样时间、农药施用时间和各自的土壤退化率(例如[12,62])。取样主要在冬季进行,通常不施用农药,但在4月初对蔬菜场地(S11)和果园(S12)进行取样。
各农业用地(S5 ~ S8和S11 ~ S14)各单项农药总浓度中位数为92 ng/g,蔬菜用地土壤农药总浓度中位数最高(S11 ~ 500 ng/g)。Riedo等[6]所研究的46种农药均属于本方法。在比较每个分析的农业土壤中量化的总浓度时,无论是基于46种农药的子集还是基于所开发方法中包含的全部146种农药,在考虑所有146种农药时,总浓度的中位数百分比增加了48%。相应的,每个农业站点的检测数量增加的中位数百分比为47%。总浓度和检测数量的明显百分比增加证实了我们的目标分析物选择及其与长期土壤监测的相关性。本文提出的用于土壤中146种农药痕量分析的多残留方法现在将应用于来自AP PPP内各种监测活动的土壤样本[19],以提供陆地暴露评估作为基础,以审查其总体目标之一,即农药施用对土壤肥力没有长期负面影响。
下载原文档:https://link.springer.com/content/pdf/10.1007/s00216-023-04872-8.pdf
为您推荐:
- 为了安全,俄罗斯限制联合国进入乌克兰核电站 2025-10-13
- 法拉·卡蒂尼的手机找到了 2025-10-13
- Sanoma的投资者页面在芬兰证券交易所晚会上获得一等奖 2025-10-13
- 新晨科技加速布局云计算 启动新一轮并购 2025-10-13
- 倡导者说,“看不见的”农村社区需要更多有针对性的心理健康支持 2025-10-13
- 由于国际货运代理业务,Tasco公布了较弱的收益 2025-10-13


